选择、填空、名词解释、简答题、论述题
自测
作业题
- 用基因工程的定义原理来阐述重组新冠病毒疫苗
- 根据你的设想设计一款转基因产品,简述它的设计思路及其应用
- 基因工程中常用工具酶种类有哪些,他们的主要作用是什么
- 什么是星星活性,引起星活性的因素有哪些,哪些措施可以抑制星活性
- 切口移位标记探针的主要步骤有哪些
- 20 μL 的酶切反应体系和 20 μL 的连接反应体系组成成分各是什么?酶切和连接的最佳反应温度又各是多少?
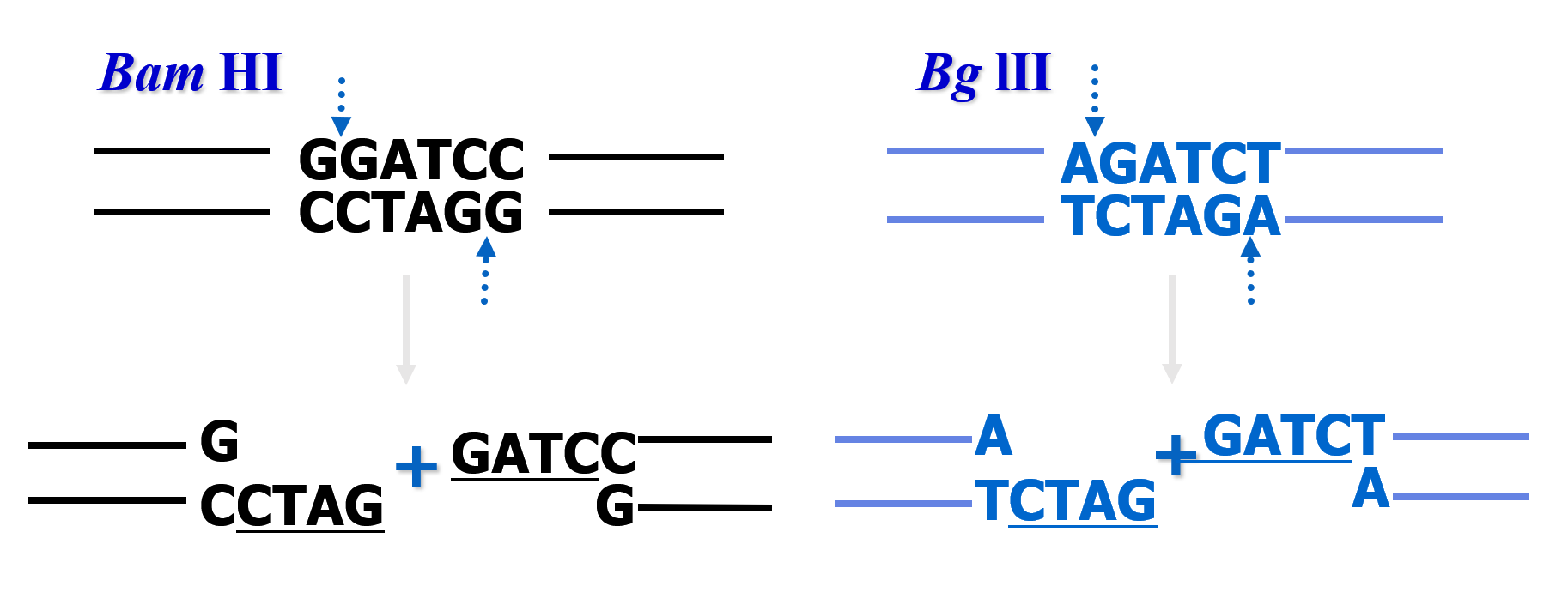
- DNA 分子平末端的连接方式有哪些?
- 理想载体至少必备的条件有哪些
- 目前的主要载体种类有哪些
- 简述蓝白斑筛选法原理
- 简述 λ 噬菌体的三种选择标记
- 简述 YAC 载体的工作原理、应用、以及其缺点
- 简述碱裂解法和煮沸法抽提质粒 DNA 的原理
- 简述电泳的基本原理
- 琼脂糖凝胶电泳过程中影响 DNA 迁移率的因素有哪些?
- 简述 PCR 的基本原理
- PCR 反应体系包含哪些成分
- 引物设计需遵循的原则有哪些
- 分子杂交的概念
- Southern 杂交的原理
- Northern 印迹与 Southern 印迹有哪些不同?
- 基因芯片的概念及应用
- 最常用的三种外源基因导入植物细胞的方法及原理
- 农杆菌 Ti 质粒的基因结构及各个结构的功能
- 简述农杆菌 Ti 质粒上的 T-DNA 导入植物基因组的过程
- 报告基因的概念
- 转植酸酶玉米,为什么要转植酸酶基因?植酸酶基因来自哪里?该转基因玉米的操作流程是怎么样的?
- 叙述 Tet-on 型四环素诱导系统的原理
- 化学降解法测序原理
- Sanger 双脱氧链终止法测序原理
- 第二代和第三代测序技术分别有哪些技术?
- 目前测序技术有哪些方面的应用
- 基因组文库和 cDNA 文库的定义、各自构建的流程、以及两者的特点
- 酵母双杂交的原理以及应用
- 动物基因工程的概念
- 转基因动物的制备方法主要有哪些?
- 简述 CRISPR/Cas9 基因定向打靶技术的原理
- 详述将基因打靶技术联合应用胚胎干细胞法制备转基因动物的流程
- 详述转基因动物技术的应用
- 酵母基因工程的优势有哪些方面?
- 详述酵母基因工程的应用进展
- 什么是基因治疗
- 目前基因药物的分类及作用原理
- 详述基因治疗在肿瘤治疗上的主要方向及重大进展
有关名词
- 基因芯片
- 基因工程
- 星星活性
- 报告基因
- 基因组 DNA 文库
- cDNA 文库
- 基因治疗
- 动物基因工程
- 同裂酶
- 同尾酶
- DNA 连接酶
- 载体
- 穿梭载体
- 质粒
- 人工染色体载体
- 黏粒载体
- 分子杂交
- 反向 PCR
其他
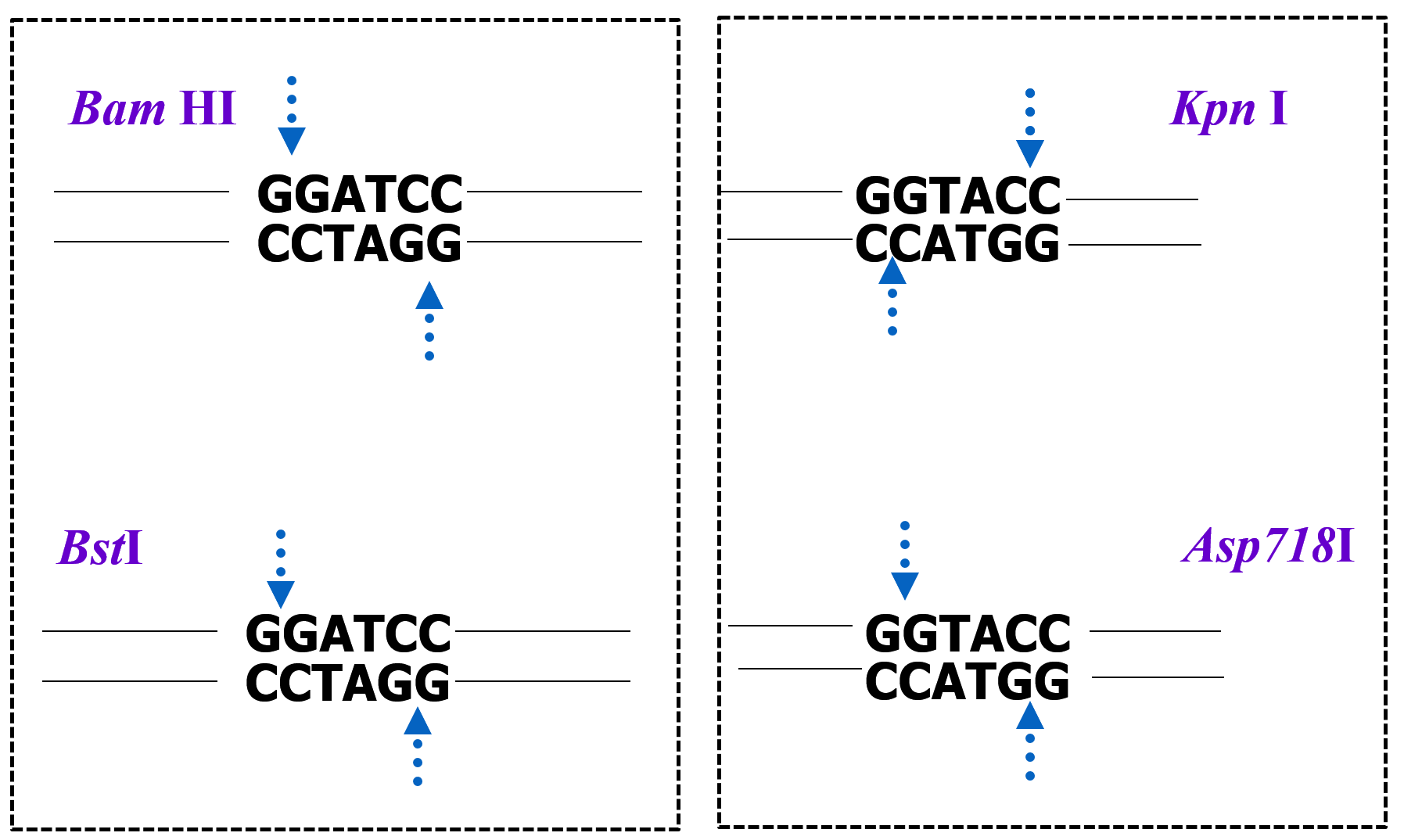
限制性核酸内切酶主要是从
下面哪项不具有限制酶识别序列的特征(
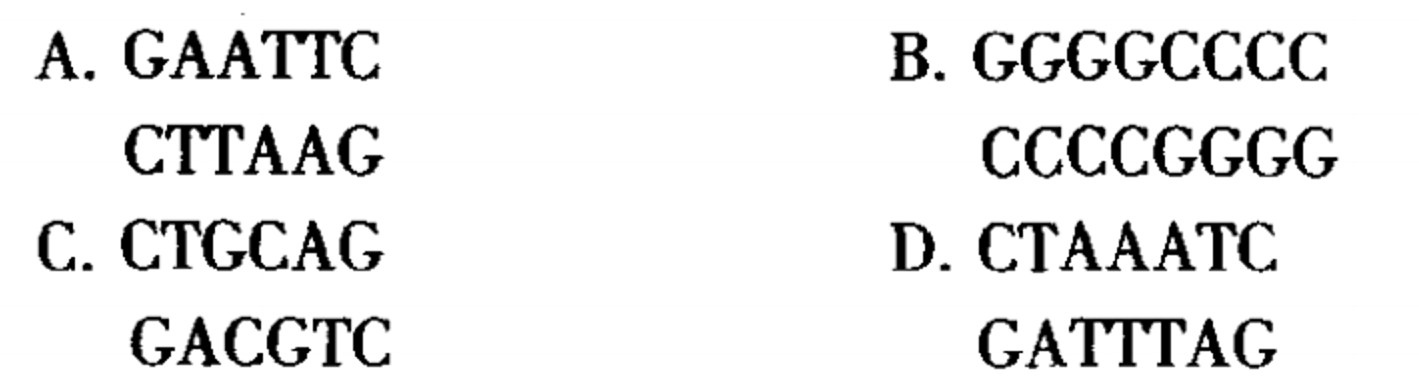
以下 粘性末端 属于 5’ 粘性末端还是 3’ 粘性末端
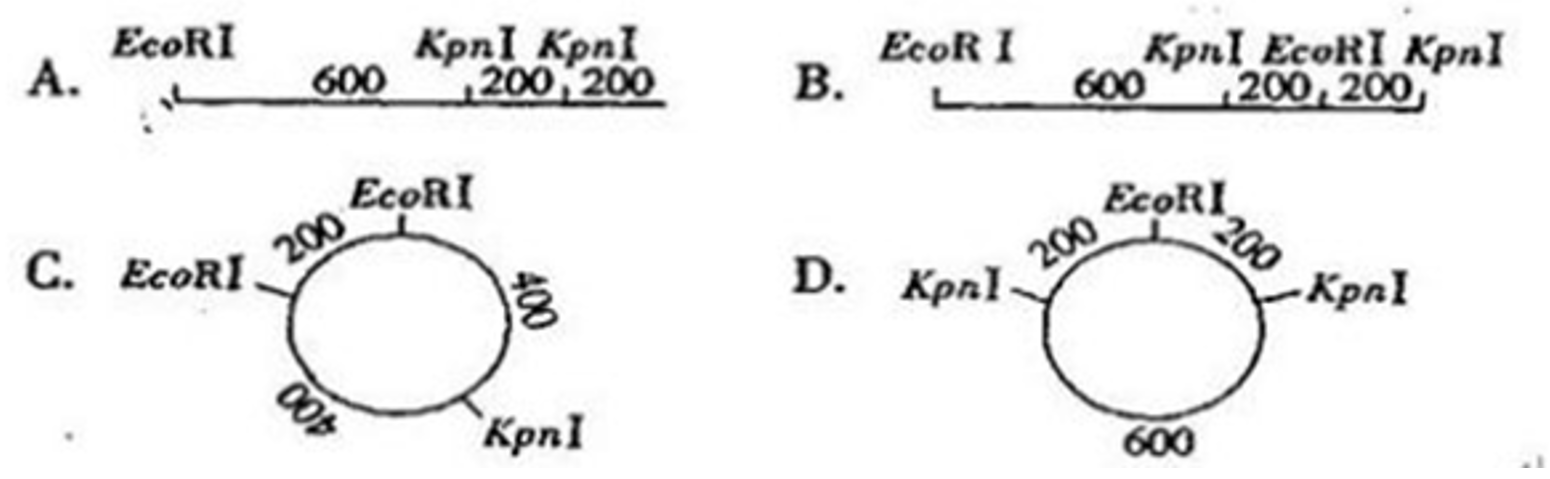
现有一长度为 1000 个碱基对(bp)的 DNA 分子,用限制性内切酶 EcoRⅠ 单独酶切后得到的 DNA 分子仍为 1000 pb,用限制性内切酶 Kpn Ⅰ 单独酶切后得到 400 pb 和 600 pb 两种长度的 DNA 分子,用 EcoR Ⅰ 和 Kpn Ⅰ 同时酶切后得到 200 pb 和 600 pb 的两种长度的 DNA 分子。该 DNA 分子的酶切图谱正确的是(
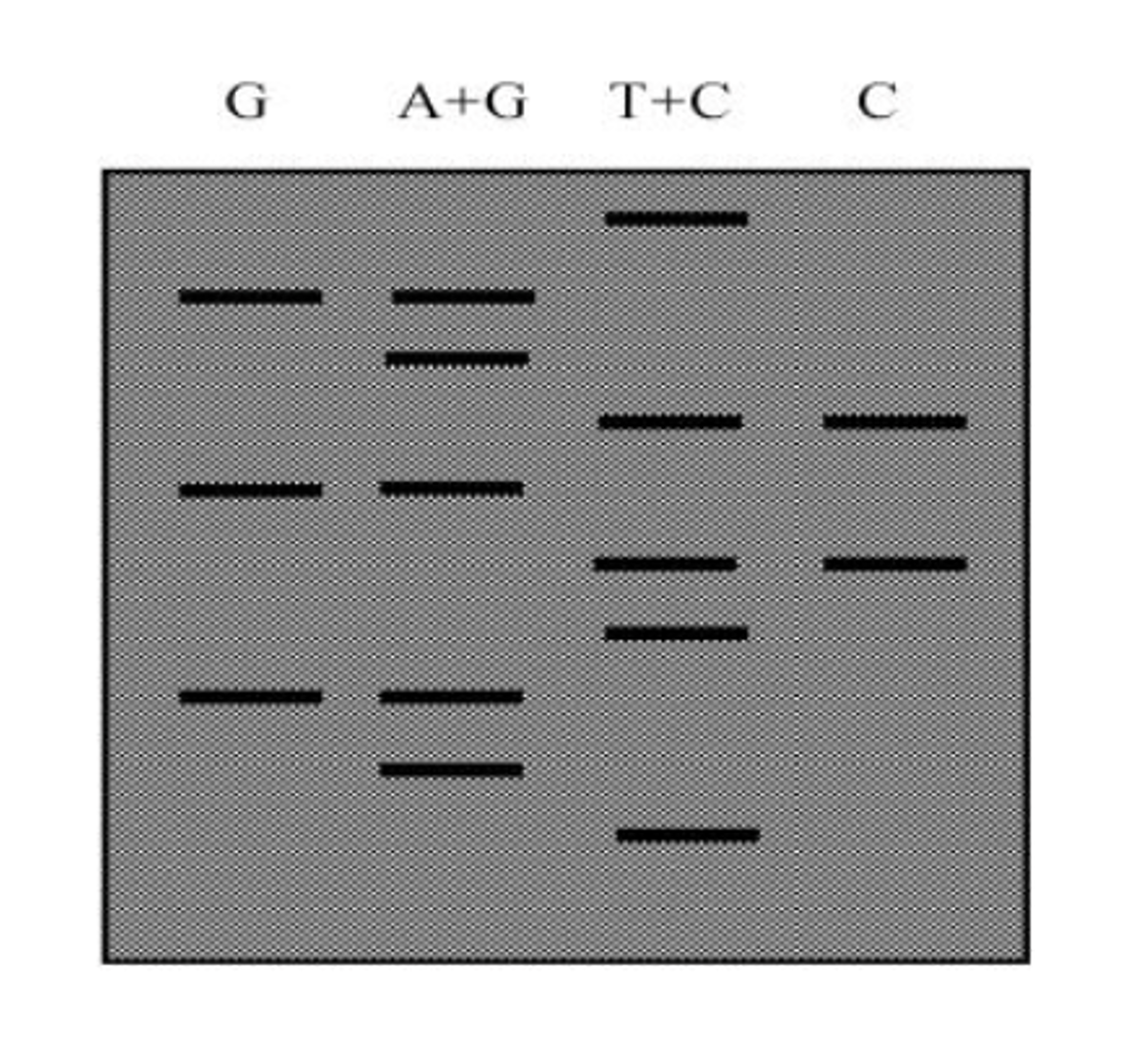
按照化学降解法测序显影的结果,读出碱基序列
按照 Sanger 双脱氧链终止法测序显影的结果,读出模板碱基序列
作业题
用基因工程的定义原理来阐述重组新冠病毒疫苗 ↩
略
重组新型冠状病毒疫苗(腺病毒载体,Ad5-nCov),将一段人工构建的保护性抗原基因整合到作为载体的病毒的基因组中,使重组后的基因片段在病毒进入宿主后被表达并产生抗体。
根据你的设想设计一款转基因产品,简述它的设计思路及其应用 ↩
略
基因工程中常用工具酶种类有哪些,他们的主要作用是什么 ↩
- 限制性内切酶:主要用于 DNA 分子的特异切割
- DNA 甲基化酶:用于 DNA 分子的甲基化
- 核酸酶:用于 DNA 和 RNA 的非特异性切割
- 核酸连接酶:用于 DNA 和 RNA 的连接
- 核酸聚合酶:用于 DNA 和 RNA 的合成
- 核酸末端修饰酶:用于 DNA 和 RNA 的末端修饰
- 其它酶类:用于生物细胞的破壁,转化,核酸纯化,检测等。
什么是星星活性,引起星活性的因素有哪些,哪些措施可以抑制星活性 ↩
在极端非标准条件下,限制酶能切割与识别序列相似的序列,这个改变的特殊性称星星活性(star activity)。↩
当酶切条件改变时,酶的专一性可能会降低,以至于同一种酶可识别和切割多个的位点。
引起星星活性的因素:
- 甘油浓度高(> 5%)
- 酶过量(> 100 U/μL)
- 离子强度低(< 25 mmol/L)
- pH 过高(> 8.0)
- 加了有机溶剂如 DMSO(二甲基亚砜)、乙醇、乙二醇、二甲基乙酰胺和二甲基甲酰胺
- 用其他 2 价阳离子如 Mn2+、Cu2+、Co2+ 或 Zn2+ 代替了 Mg2+
但不同的酶对上述条件的敏感性不一样,如 Pst Ⅰ 比 Eco R Ⅰ对高 pH 更敏感,但后者对甘油浓度更敏感。
抑制星星活性的措施:
- 减少甘油浓度;
- 减少酶的用量(可避免过分酶切);
- 提高离子强度到 100 ~ 150 mmol/L(如果不会抑制酶的活性的话);
- 降低反应 pH 至 pH 7.0 ;
- 保证反应体系中无有机溶剂或乙醇;
- 保证使用 Mg2+作为 2 价阳离子。
切口移位标记探针的主要步骤有哪些 ↩
P33
利用大肠杆菌 DNA 聚合酶Ⅰ的 5’ → 3’ 外切核酸酶活性,可用切口平移法(nick translation)标记 DNA。
- 首先在镁离子存在下用低限量的 DNaseⅠ 处理双链 DNA 模板,产生少量切口
- 在切口处,利用大肠杆菌 DNA 聚合酶Ⅰ的 5’ → 3’ 外切核酸酶活性使切口沿 5’ → 3’ 方向平移,同时产生切口也可作为大肠杆菌 DNA 聚合酶Ⅰ催化 DNA 合成的引物
- 合成过程中,dNTP 前体不断掺入到正在 DNA 链上。如果提供标记的 dNTP,那么所有的反应产物便被标记,可用作杂交探针。
20 μL 的酶切反应体系和 20 μL 的连接反应体系组成成分各是什么?酶切和连接的最佳反应温度又各是多少? ↩
20 μL 的酶切反应体系:
| 总体积 | 20 μL |
|---|---|
| DNA | 1 μL(1 μg) |
| buffer(10x) | 2 μL |
| ddH2O | 16 μL |
| 限制性内切酶 | 1 μL |
反应温度:不同的限制性内切酶的最适反应温度不同。大多数 37℃,少数要求 40℃ ~ 65℃
20 μL 的连接反应体系,以 T4 DNA 连接酶的反应为例:
| 总体积 | 20 μL |
|---|---|
| 10× T4 DNA Ligase Buffer | 2 μL |
| DNA 片段 | 约 0.3 pmol |
| 载体 DNA | 约 0.03 pmol |
| T4 DNA Ligase | 1 μL |
| ddH2O | up to 20 μL |
反应温度:16 ℃
DNA 片段的摩尔数应控制在载体 DNA 摩尔数的 3 ~ 10 倍
DNA 分子平末端的连接方式有哪些? ↩
- 直接用 T4 DNA 连接酶连接;
- 先用末端核苷酸转移酶,给平末端 DNA 分子加上同聚物尾巴之后再用 DNA 连接酶进行连接;
- DNA 接头连接法:它是一类由人工合成的一头具有某种限制性内切酶粘末端,另一头为平末端的特殊的双链寡核苷酸片段。当它的平末端与平末端的外源 DNA 片段连接之后,便会使后者成为具黏性末端的新的 DNA 分子,而易于连接重组。
理想载体至少必备的条件有哪些 ↩
- 质粒拷贝数较高。质粒拷贝数是指生长在标准的培养基条件下,每个细菌细胞中所含有的质粒 DNA 分子的数目。根据宿主细胞所含的拷贝数多少,可将质粒分成
- 严谨型:低拷贝数的质粒,每个宿主细胞中仅含有 1 ~ 2 份的拷贝
- 松弛型:高拷贝数的质粒,每个宿主细胞中可高达 10 ~ 200 份拷贝
- 分子量较小。低分子量的质粒如下优点:
- 外源 DNA 容量较大
- 容易转化,当质粒大于 15 kb 时,将成为转化效率的制约因素
- 遗传工程操作时容易拿捏,容易分离,不易断裂
- 带有可供标记基因。选择标记是指带有一种或数种抗生素抗性基因,使寄主获得同样的抗生素抗性性状(resistance)。抗生素选择基因有:氨苄青霉素(Amp),氯霉素(Cm),四环素(Tet)和卡那霉素(Kan)四种,而且希望各抗性基因内有若干单一的限制酶切点。
- 带有尽可能多的单一限制性酶切位点。单一的限制性酶切位点可供外源 DNA 定点插入,目前常用载体上的多克隆位点(multiple cloning sites, MCS)即具有该功能。
- 具有复制起始点(origin, ori)。这是质粒自我增殖所必不可少的基本条件,也是决定质粒拷贝数的重要元件,可使繁殖后的细胞维持一定数量的质粒拷贝数。
目前的主要载体种类有哪些 ↩
- 克隆载体(cloning vector):主要是对目的基因克隆,建立 DNA 文库和 cDNA 文库,其上有复制子即可;
- 表达载体(expression vector):能使目的基因在宿主细胞中表达的一类载体。这类载体既有复制子,更要有强启动子;
- 穿梭载体(shuttle vector):这类载体可以在原核细胞中复制,也可在真核细胞中扩增和表达。(2 种复制子和 2 种标记基因)↩
- 质粒载体
- 病毒载体(细菌、植物、动物)
- 人工染色体载体
简述蓝白斑筛选法原理 ↩
P44
- 由 α-互补而产生的 LacZ + 细菌在诱导剂 IPTG(异丙基硫代半乳糖苷)的作用下,在底物 X-gal 存在时产生蓝色菌落。
- 而当外源 DNA 插入到质粒的多克隆位点(MCS)后,α-互补被插入失活,使得带有重组质粒的 LacZ - 细菌形成白色菌落。
β-半乳糖苷酶可以拆分成两个部分,N 端和 C 端。β-半乳糖苷酶缺陷型菌株的基因组中含有表达 β-半乳糖苷酶 C 端的基因,而 N 端(一个 146 个氨基酸的短肽,即 α 肽链)的基因被安放到了表达的载体中。N 端基因经过改造,中间插入多克隆位点。这段经过改造的 N 端基因被称为 lacZ 基因
lacZ 基因是乳糖 lac 操纵子中编码 β-半乳糖苷酶的基因,乳糖及其衍生物可诱导其表达。
α-互补是指 β-半乳糖苷酶的两个无活性片段(N 端片段和 C 端片段)组合成为功能完整的酶的过程,指 lacZ 基因上缺失近操作基因区段的突变体与带有完整的近操作基因区段的 β-半乳糖苷酶基因的突变体之间实现互补。大肠杆菌 β-半乳糖苷酶可以和其底物 X-gal 相互作用并且释放出一种蓝色物质,当该酶的 α-片段和 ω-片段分开时就失去了这种显色的功能。通常将编码该酶 α-片段的 LacZ ’ 基因插入到载体的多克隆位点的侧翼序列中,而在一些人工构建的大肠杆菌株系中却只能编码产生该酶的 ω 片段。这样一来,含有功能性完整的 LacZ ’ 基因的载体导入到这类寄主细胞中时,载体编码的 α-片段就能和寄主编码的 ω-片段发生互补并具有了对底物 X-gal 的作用功能(发生显色反应),这种现象被成为是 α-互补。
缺陷株基因组无法单独编码有活性的 β-半乳糖苷酶,但当菌体中含有带 lacZ 的质粒后,质粒 lacZ 基因编码的 α 肽链(酶的 N 端)和菌株基因组表达的 β-半乳糖苷酶的 C 端互补,具有与完整 β-半乳糖苷酶相同的作用,具有分解 X-gal 生成蓝色物质的能力。操作中,添加 IPTG 以激活 lacZ 中的 β-半乳糖苷酶的启动子,在含有 X-gal 的固体平板培养基中菌落就会呈现蓝色。但是当 MCS 位点上插入 DNA 片段时,lacZ 基因就失活了,没法完成 α-互补,β-半乳糖苷酶失活,无法分解 X-gal,因此菌落就是白色的。
简述 λ 噬菌体的三种选择标记 ↩
P54
在 λ 基因组中引入 LacZ ’ 序列(其上含有一个 EcoRⅠ克隆位点)。感染 LacZ ’ 突变的大肠杆菌,经 IPTG 的诱导,利用 X-gal 的显色反应作选择标记
cⅠ基因的表达促进 λ 噬菌体进入溶原状态,插入导致载体不能合成阻遏物,λ 载体 DNA 不能进入溶源期,受体菌裂解,形成清晰的噬菌斑。没有外源 DNA 插入的 λ 载体感染受体菌形成混浊噬菌斑。
- 野生型 λ 噬菌体不能在携带有 P2 原噬菌体的溶源菌中生长, λ 的这种表型称为Spi+(对 P2 的干扰敏感)。
- 当 λ 基因组中缺少参与重组的 red 和 gam 基因时, λ 突变体便可以在 P2 溶原菌中生长,其表型称为 Spi-。
- 基因 red 和 gam 位于非必需区域内,外源 DNA 片段能置换基因 red 和 gam 形成重组体,其重组体能在 recA + 的 P2 溶原菌中繁殖,并且显示出 Spi- 的表型。
- 因此只要选用 recA + 的 P2 溶原菌作为宿主菌,就可以对重组体进行筛选。能浸染宿主菌产生噬菌斑的即是重组分子。
简述 YAC 载体的工作原理、应用、以及其缺点 ↩
P81
工作原理:对于 BamHⅠ 切割后形成的微型酵母染色体,当用 EcoRⅠ 或 Sma Ⅰ 切割抑制基因 sup 4 内部的位点后形成染色体的两条臂,与外源大片段 DNA 在该切点相连就形成一个大型人工酵母染色体,转化到酵母菌后可像染色体一样复制,并随细胞分裂分配到子细胞中去,达到克隆大片段 DNA 的目的。外源 DNA 片段的装载导致抑制基因 sup 4 插入失活,从而使重组菌形成红色菌落;而载体自身连接转入到酵母细胞后所形成的菌落为白色。
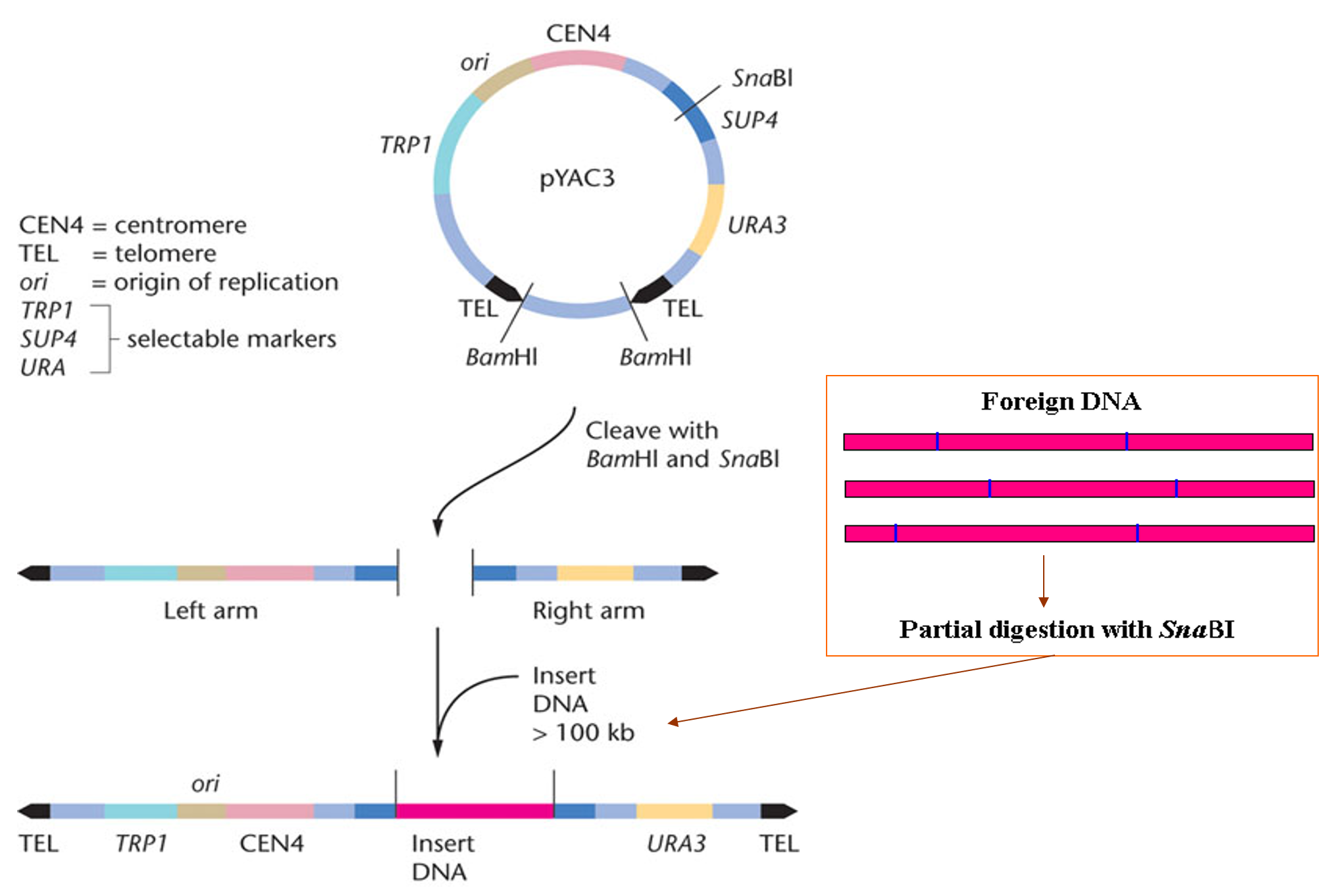
YAC 的主要用途:
- YAC 克隆重叠群是物理图谱的主要框架
- 可容纳大片段(250 ~ 400 kb)的外源 DNA,在构建基因组文库时需要较少的克隆
- 在人类、植物、昆虫、鸟类、两栖类等复杂的生物基因组分析和 DNA 测序中发挥着重要作用。相比于早期 DNA 载体,YAC 是容纳大片段外源 DNA 的首选载体,成为人类基因组计划及图位克隆分离基因的重要工具,并促进了发展人类人工染色体(HAC)的研究
- 基因功能研究中,基因嵌入及转基因技术都采用 YAC 克隆系统。通过 YAC 嵌入的基因不仅片段长,而且嵌入的基因更适于体内表达,并有利于基因保持其固有的空间构象和功能的发挥
YAC 的缺点:
- 插入片段大,稳定性较差,不易操作
- 插入的大片段常发生缺失,使文库不完整
- YAC 与酵母天然染色体分子结构相似,分离时难与天然染色体分开
- 文库中的嵌合现象严重
- 因插入片段大,往往发生序列重排,造成序列错乱
简述碱裂解法和煮沸法抽提质粒 DNA 的原理。 ↩
染色体 DNA 比质粒 DNA 分子大得多,且染色体 DNA 为线状分子,而质粒 DNA 为共价闭合环状分子;
当用碱处理 DNA 溶液时,线状染色体 DNA 容易发生变性,共价闭环的质粒 DNA 再回到中性 pH 时即恢复其天然构象;
当加热处理 DNA 溶液时,线状染色体 DNA 容易发生变性,共价闭环的质粒 DNA 在冷却时即恢复其天然构象;
变性染色体 DNA 片段与变性蛋白质和细胞碎片结合形成沉淀,而复性的超螺旋质粒 DNA 分子则以溶解状态存在液相中,从而可通过离心将两者分开。
简述电泳的基本原理。 ↩
带电粒子可以用电泳技术进行分离、分析和鉴定的基本原理:在一定 pH 条件下,每一种分子都具有特定的电荷(种类和数量)、大小和形状,在一定时间内它们在相同电场中泳动速度不同,各自集中到特定的位置上而形成紧密的泳动带。
琼脂糖凝胶电泳过程中影响 DNA 迁移率的因素有哪些 ↩
影响 DNA 迁移率的因素:
DNA 分子的大小、构象,凝胶浓度,电压,电泳缓冲液的组成等。
- DNA 带负电荷,迁移率与分子量对数成反比,DNA 片段越长,泳动速度越慢
- DNA 分子构象影响迁移率:共价闭环 DNA > 线状 DNA > 开环 DNA
- 泳动速度与凝胶浓度成反比
- 在低电压时,泳动速度与电场强度成正比
- 电泳缓冲液的组成及其离子强度会影响 DNA 的电泳迁移率
简述 PCR 的基本原理 ↩
- 由一对引物介导,通过温度的调节,使双链 DNA 变性为单链 DNA、单链 DNA 与引物复性(退火)成为引物-DNA 单链复合物、以及在 dNTPs 存在下 DNA 聚合酶使引物延伸而成为双链 DNA(引物的延伸)
- 这种热变性-复性-延伸的过程,就是一个 PCR 循环
- 一般通过 20 ~ 30 个循环之后,就可获得大量的要扩增的 DNA 片段。
PCR 反应体系包含哪些成分 ↩
PCR 反应体系包含:
- 模板 DNA
- Taq 酶(Taq DNA polymerase)
- dNTP
- 引物(Primers)(上游、下游)
- 缓冲液体系(buffer)
- Mg2+
- ddH2O
引物设计需遵循的原则有哪些 ↩
- 引物长度以 18 ~ 30 个碱基为宜,过长过短都会降低特异性
- G+C 的含量一般为 40% ~ 60%,提高引物与模板的结合力
- Tm(解链温度)要合适,正反两引物间相差不大于 5℃
- 四种碱基随机分布,避免连续相同碱基排列
- 引物自身不应存在互补序列,以防形成发夹结构
- 两个引物之间不应有多于 4 个的互补,尤其避免 3’ 端的碱基互补,以免形成引物二聚体
- 引物 5’ 末端碱基可不与模板 DNA 紧密匹配,可添加与模板无关的序列(如限制性核酸内切酶),便于 PCR 产物的定向克隆。5’ 端是可修饰的。
- 引物 3’ 末端碱基一般应与模板 DNA 严格配对,并且 3’ 末端为 G 或 C 时引发效率较高,但不要 GC 连排。引物的延伸是在 3’ 端开始,3’ 端不能进行任何修饰
- 引物的碱基顺序不能与非扩增区有同源性,即引物要有特异性
分子杂交的概念 ↩
分子杂交:是一类核酸和蛋白质分析方法,用于检测混合样品中特定核酸分子或蛋白质分子是否存在,以及其分子量的大小。
根据其检测对象不同,可分为:
| 分子杂交 | 检测对象 | 探针 |
|---|---|---|
| Southern 杂交 | DNA | 核酸 |
| Northern 杂交 | RNA | 核酸 |
| Western 杂交 | 蛋白质 | 抗体 |
以及由此而简化的斑点杂交、菌落杂交和原位杂交等。
Southern 杂交的原理 ↩
通过毛细管作用、电转移、真空转膜等方法,使在凝胶电泳中已分离的 DNA 片段转移并结合到适当的滤膜上,然后通过同已标记的探针进行杂交,以检测被转移 DNA 片段,称为 DNA 印迹杂交技术。
Northern 印迹与 Southern 印迹有哪些不同 ↩
- 转移的对象不同,Northern 印迹是将 RNA 变性及电泳分离后,将其转移到固相支持物上的过程。
- 虽然 RNA 电泳前不需像 DNA 那样进行酶切,但也需要变性。不过变性方法是不同的,它不能用碱变性,因为碱变性会导致 RNA 的降解,RNA 变性(解二级结构)一般使用甲醛
基因芯片的概念及应用 ↩
基因芯片:是一种高通量的斑点杂交技术,通过将大量的 DNA 分子固定于支持物上,并与标记的样品杂交,然后通过自动化仪器检测杂交信号来判断样品中靶分子的信息,诸如基因组 DNA 突变谱和 mRNA 表达谱的检测等。↩
基因芯片的应用:
- 表达谱分析
- SNP 分析(包括最新的 CSVI/CNV 分析,GWAS)
- CGH 分析
- ChIP-chip 分析
- 传染性疾病的检测(有无和耐药性检测)
- 遗传性疾病的检测
最常用的三种外源基因导入植物细胞的方法及原理 ↩
以原生质体为受体,借助于特定的化学或物理手段将外源 DNA 直接导入植物细胞的方法。有 PEG 介导的基因转化、脂质体介导的基因转化、电激法介导的基因转化、显微注射法介导的基因转化、激光微束介导的基因转化等。
将外源 DNA 包被在微小的金粒或钨粒表面,然后在高压的作用下将微粒高速射入受体细胞或组织。微粒上的外源 DNA 进入细胞后,整合到植物染色体上并得到表达,从而实现外源基因的转化。
根癌农杆菌感染植物,其中 Ti 质粒中的一部分 DNA 片段进入寄主细胞并插入基因组中。T-DNA 中基因利用植物的酶系统进行转录和翻译
农杆菌 Ti 质粒的基因结构及各个结构的功能 ↩
- T-DNA 区:转移到植物基因组的一段 DNA
- Vir 区(毒性区):激活 T-DNA 转移,使植物致瘤
- Ori 区:质粒复制起点
- Con 区:调控 Ti 质粒在根癌农杆菌间的转移
简述农杆菌 Ti 质粒上的 T-DNA 导入植物基因组的过程 ↩
P269
农杆菌 Ti 质粒上的 T-DNA 导入植物基因组整个过程大致可分为以下 6 个步骤:
- 农杆菌对受体的识别
- 农杆菌附着到植物受体细胞
- 诱导启动毒性区基因表达
- 类似接合孔复合体的合成和装配
- T-DNA 的加工和转运
- T-DNA 的整合
报告基因的概念 ↩
报告基因是指一类在细胞、组织、器官或个体处于特定情况下会表达并使得他们产生易于检测、且实验材料原本不会产生的性状的基因。
作为报告基因,在遗传选择和筛选检测方面须具有以下条件:
- 已被克隆和全序列已测定;
- 表达产物在受体细胞中本不存在,即无背景,在被转染的细胞中无相似的内源性表达产物,且不损坏受体细胞。
- 其表达产物能进行定量测定
转植酸酶玉米,为什么要转植酸酶基因?植酸酶基因来自哪里?该转基因玉米的操作流程是怎么样的? ↩
植酸酶是催化植酸及其盐类水解为肌醇与磷酸(盐)的一类酶的总称,具有特殊的空间结构,能够依次分离植酸分子中的磷,将植酸(盐)降解为肌醇和无机磷,同时释放出植酸(盐)结合的其他营养物质。
猪和禽类等单胃动物因缺乏分解植酸的酶而难以利用植酸,造成有效磷利用率低和环境污染等问题,因此通过转基因技术提高玉米中植酸酶含量,改善玉米饲料的品质,对节约成本、提高饲料利用率及保护环境等有重要的意义。
植酸酶广泛存在于自然界中,植物、部分动物组织和微生物都存在着植酸酶,主要来源有黑曲霉。
转基因玉米的操作流程:
- 获得植酸酶基因,克隆到载体上扩增
- 构建植物基因转化受体系统
- 目的基因导入受体细胞,例如通过基因枪法、花粉管通道法将外源植酸酶基因导入受体
- 培养转化细胞
- 筛选、培植、鉴定。进行转基因植株检测,有 PCR、RT-PCR、Western 植酸酶活性等检测
叙述 Tet-on 型四环素诱导系统的原理。 ↩
Tet(四环素)诱导调控表达系统的基本原理是由诱导药物如 Tet 改变调控蛋白质的构象,从而控制目标蛋白质的表达最初的 Tet 诱导调控基因表达系统是以大肠杆菌 Tn10 转座子上 Tet 抗性操纵子为基础而建立的。Tet 阻遏蛋白(Tet repressor protein, TetR)与 Tet 操纵基因(Tet operator, TetO)DNA 序列有特异的亲和能力,当细胞内无 Tet 存在时,TetR 会与 TetO 结合,从而阻断下游的抗性基因表达。当有 Tet 存在时,药物使 TetR 的构象发生改变,导致 TetR 与 TetO 分离,从而引起抗性基因的抑制解除,抗性蛋白表达使细菌产生耐药性。利用 TetR 和 TetO 特异结合的特性,研究人员发展了多种类型的 Tet 调控系统,但根据其表达特点可以归为两大类:抑制型系统 Tet-off 和激活性系统 Tet-on。

Tet 系统的基本结构示意图。Tet 系统由调控蛋白(tTA, rtTA, tTS, rtTS)的表达框和目的基因表达框组成。调控蛋白由一个强启动子驱动;而在目的基因的表达框中,启动子位于 Tet 应答元件(Tet-responsive element, TRE)的下游,调控蛋白可以通过 TRE 对下游启动子进行调控。
(一)Tet-off 系统
Gossen 等最早建立了 Tet-off 基因调控表达系统,该系统由调节表达载体和反应表达载体组成。调节表达载体包含一个由人巨细胞病毒早期启动子(PhCMV)启动的 Tet 转录活化因子(tetracycline transcriptional activator, tTA),tTA 由 TetR 与单纯疱疹病毒(HSV)VP16 蛋白质 C 端的一段转录激活区融合而成。反应表达载体由 Tet 应答元件(Tet-responsive element, TRE)、最小 CMV 启动子(minimal CMV promoter, PminCMV)及目的基因组成。目的基因位于 TRE 和 PminCMV 的下游;TRE 为 7 次重复的 TetO 序列。PminCMV 缺失增强子,因此在 tTA 未结合到 TRE 时,目的基因不表达;相反,当 tTA 结合到 TRE 时,VP16 会使 PminCMV 活化从而使基因表达。当细胞内无 Tet 或其衍生物强力霉素(doxcycline, Dox)的存在时,tTA 可与 TRE 结合,打开基因表达;而当Tet 或 Dox 存在时,它们可使 tTA 中的 TetR 改变构象,则 tTA 将从 TRE 上脱落下来,使 TRE 中的 PminCMV 处于非激活状态,从而使基因表达处于关闭状态。

Tet-off 示意图。tTA 是由 TetR 和病毒转录激活域 VP16 融合而成的蛋白。当不存在 Dox 时,tTA 和 TRE 结合,启动 PminCMV 的下游基因表达;当存在 Dox 时,Dox 使 tTA 的构象发生改变, tTA 会从 TRE 上脱落,从而导致 TRE-PminCMV 下游基因表达关闭。
(二)Tet-on 调控系统
Tet-on 调控系统与 Tet-off 调控系统的区别在于其调节蛋白质为反义 Tet 转录活化因子(reverse tetracycline transcriptional activator, rtTA)。rtTA 是由反义 TetR(reverse TetR, rTetR)与 VP16 的转录活化区域融合而成。rTetR 由 TetR 中的 4 个氨基酸发生突变而衍生而来的(E71 → K71, D95 → N95, L101 → S101, G102 → D102)。rTetR 的表型与 TetR 相反,在无 Dox 时其不能结合 TRE,导致基因表达关闭;而在有 Dox 存在时其会结合在 TRE 上,导致基因表达开放

Tet-on 示意图。rtTA 是由 rTetR 和 VP16 融合而成的蛋白,它的表型与 tTA 相反。当 Dox 不存在时,rtTA 不能结合 TRE, TRE-PminCMV 下游基因表达关闭;当存在 Dox 时,rtTA 构象改变,rtTA 结合 TRE,则 TRE-PminCMV 下游基因表达开启。
陈皓,夏海滨.四环素诱导调控表达系统的研究与应用[J].生命的化学,2011,31(02):285-291.
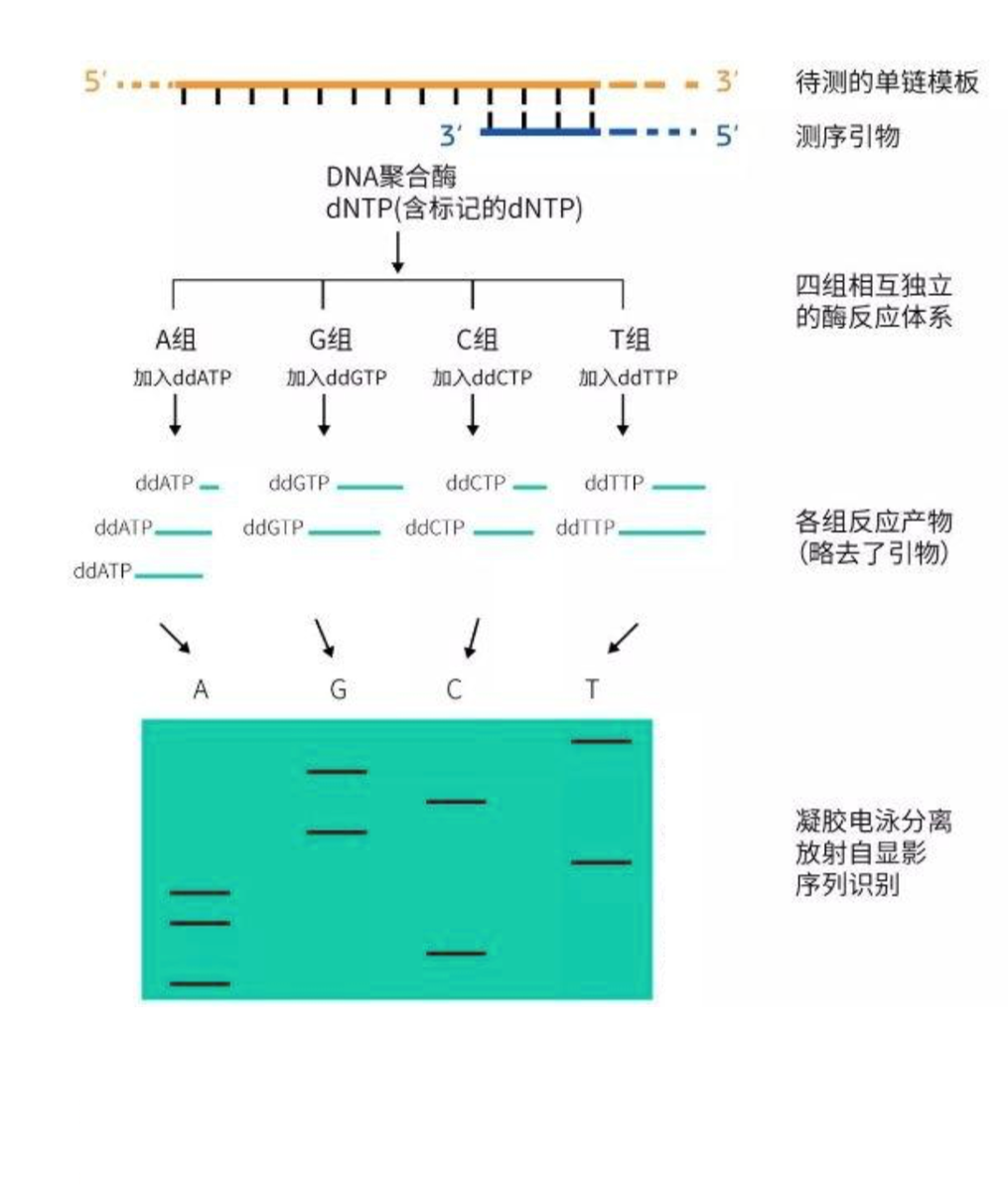
化学降解法测序原理 ↩
将一个 DNA 片段的 5’ 端磷酸基作放射性标记,再分别采用不同的化学方法修饰和裂解特定碱基,打断磷酸二脂键,从而产生一系列长度不一而 5’ 端被标记的 DNA 片段,这些以特定碱基结尾的片段群通过凝胶电泳分离,再经放射线自显影,确定各片段末端碱基,从而得出目的 DNA 的碱基序列。
Sanger 双脱氧链终止法测序原理 ↩
P167
双脱氧链终止测序方法巧妙地使用了双脱氧核苷酸(2’,3’-ddNTP,N 指 A,T,G 或 C)在同一扩增(-OH),它与正常情况下合成 DNA 的脱氧核苷酸(2’-dNTP)的主要不同点在于 3’ 位置的羟基缺失。当它与正常核苷酸混合于同一个扩增反应体系中,在 DNA 聚合酶的作用下,虽然它也能够像 2’-脱氧核苷酸一样参与 DNA 合成,以其 5’ 位置的磷酸基团与上位脱氧核苷酸的 3’ 位置的羟基结合;但是,由于它自身 3’ 位置的羟基缺失,至使下位核苷酸的 5’ 磷酸基无法与之结合,使得正在合成的 DNA 链的合成反应终止于此双脱氧核苷酸。
基于双脱氧核苷酸的这种特性,Sanger 于 1977 年建立了以双脱氧终止反应为基础的 DNA 序列测定的方法。该方法以待测单链或双链 DNA 为模板,使用能与 DNA 模板结合的一段寡核苷酸为引物,在 DNA 聚合酶的催化作用下合成新的 DNA 链。正常情况下的 DNA 聚合酶催化反应在其反应体系中包含 4 种脱氧核苷酸(dATP、dCTP、dGTP 和 dTTP),合成与模板 DNA 互补的新链。当向这个反应体系中加入一种双脱氧核苷酸(ddATP、ddCTP、ddGTP 和 ddTTP)后,在 DNA 合成过程中,ddNTP 将与相应的 dNTP 竞争掺入到新合成的 DNA 互补链中。
- 如果 dNTP 掺入其中,DNA 互补链则将继续延伸下去;
- 而如果 ddNTP 掺入其中,DNA互补链的合成则到此终止。
按照这一反应方式,可得到 4 种分别以 ddATP、ddCTP、ddGTP 和 ddTTP 结尾的不同长度 DNA 片段群。
由于反应时新生 DNA 片段的长度取决于模板 DNA 中与该双脱氧核苷酸相对应的互补碱基的位置,即双脱氧核苷酸掺入的位置,而双脱氧核苷酸的掺入是随机的,故各个新生 DNA 片段的长度互不相同。不同长度 DNA 片段在凝胶中的移动速率不同,而聚丙烯酰胺凝胶电泳分辨率极高,能分辨出小至 1 个碱基长度差的 DNA 片段,从而将混合产物中不同长度 DNA 片段分离开,再通过放射自显影曝光,根据片段尾部的双脱氧核苷酸读出该 DNA 的碱基排列顺序。
第二代和第三代测序技术分别有哪些技术? ↩
- 罗氏 454 公司的 GS FLX 测序平台
- Illumina 公司的 Solexa Genome Analyzer 测序平台
- ABI 公司的 SOLiD 测序平台
- Life Techologies 公司的离子肼测序
- Helicos 公司的 Heliscope 单分子测序仪
- Pacific Biosciences 公司的 SMRT 技术
- Oxford Nanopore Technologies 公司正在研究的纳米孔单分子技术
目前测序技术有哪些方面的应用? ↩
- 用于科研,建立基因序列数据库
- 用于临床,如肿瘤基因的检测、生育健康、病原微生物的检测、测序分析等
- 用于生物资源的保存
- 用于亲缘分析
仅作参考
基因组文库和 cDNA 文库的定义、各自构建的流程、以及两者的特点 ↩
基因文库(Gene library):由某一生物的全部或部分基因的不同 DNA 片段的克隆所构成的群体, 称之为基因文库。一个完全的基因文库,应该能够保证从中筛选到目的基因。
基因组 DNA 文库:指将某生物体的全部基因组 DNA 用限制性内切酶或机械力量切割成一定长度范围的 DNA 片段,与合适的载体体外重组并转化相应的宿主细胞获得的所有阳性菌落。 包含基因的全部信息,如编码区,非编码区,内含子和外显子、启动子及调控序列。↩
cDNA 文库:是指将某种生物体基因组转录的全部 mRNA 经反转录产生的 cDNA 片段分别与克隆载体重组,储存于某种受体菌中,该群体就称该生物基因组的 cDNA 文库。↩
基因组 DNA 文库的构建程序:
- 载体的制备;
- 高纯度大分子量基因组 DNA(HMW DNA)的提取;
- HMW DNA 的部分酶切与脉冲电泳分级分离(PFGE size selection);
- 载体与外源片段的连接与转化或侵染宿主细胞;
- 重组克隆的挑取
- 文库的保存。
cDNA 文库的构建主要分为四步:
- 细胞总 RNA 的提取和 mRNA 分离
- 第一链 cDNA 合成;
- 第二链 cDNA 合成;
- 双链 cDNA 克隆进质粒或噬菌体载体并导入宿主中繁殖
- cDNA 克隆只能反映 mRNA 的分子结构,没有包括基因组的间隔序列,并且受细胞来源或发育时期的影响。
- cDNA 文库中,不同克隆的分布状态总是反映 mRNA 的分布状态,即:
- 高丰度 mRNA 的 cDNA 克隆,所占比例较高,分离基因容易;
- 低丰度 mRNA 的 cDNA 克隆,所占比例较低,分离基因困难;
- 从 cDNA 克隆中,不能克隆到基因组 DNA 中的非转录区段序列,不能用于研究基因编码区外侧调控序列的结构与功能。
- cDNA 文库以 mRNA 为材料,特别适用于某些 RNA 病毒等的基因组结构研究及有关基因的克隆分离。
- cDNA 文库的筛选比较简单易行。
- 每一个 cDNA 文库都含有 mRNA 序列,这样在目的基因的选择中出现假阳性的概率就会比较低,因此阳性杂交信号一般都是有意义的,由此选择出来的阳性克隆将会含有目的基因。
- cDNA 克隆还可用于真核细胞 mRNA 的结构和功能研究。
酵母双杂交的原理以及应用 ↩
《基因工程》P226
双杂交系统的基本原理来自对酵母转录激活因子(transcriptional activator)GAL4 的认识。许多真核生物转录激活因子有两个功能域:
- 一是 DNA 结合结构域(DNA binding domain,BD),可与 DNA 序列的特定位点即上游激活序列(upstream activating sequence,UAS)结合;
- 二是转录激活结构域(activation domain,AD),协助 RNA 聚合酶Ⅱ复合体激活 UAS 下游基因的转录。
这两个结构域的功能是独立的。正常情况下它们都是同一种蛋白质的组成部分,缺一不可,但如果利用 DNA 重组技术把它们彼此分开并放置在同一宿主中表达,也不能激活相关基因的转录,其原因是由于它们彼此之间在空间上存在一定距离,不会直接发生相互作用。如果能将它们空间上的距离拉近,就可以形成有功能的转录激活因子,从而启动下游基因的转录。
酵母双杂交理论就是建立在此原理上,利用融合蛋白的策略,将蛋白 X 与 BD 融合,蛋白 Y 与 AD 融合,将它们导入酵母细胞中共表达,如果 X 与 Y 相互作用,则会导致 BD 和 AD 在空间上接近,形成一个有功能的转录激活因子,激活下游报告基因的表达。
- X 为已知蛋白,把要筛选的“探针 X”与 BD 构建融合蛋白 BD-X(通常称为诱饵,bait),并以含有 BD-X 融合蛋白基因和报告基因的细胞为构建文库的受体菌;
- 而将所要筛选的对象 Y 与 AD 构建成融合蛋白 AD-Y 的 cDNA 文库(即将目的 cDNA 与 AD 基因构建融合基因文库),通常称 AD-Y 为猎物(prey),当 Y 基因产物能与 X 基因产物发生作用时,就可启动报告基因的表达。

A. GAL4 的 BD 和蛋白质 X 形成的融合蛋白同 GAL1 的 UAS 序列结合,但由于没有 AD 的结合所以不能激活报告基因的转录;
B. GAL4 的 AD 与蛋白质 Y 形成的融合蛋白没有 BD 结合时,也不能激活报告基因的转录;
C. BD-X 和 AD-Y 的相互作用重建了 GAL4 的功能,使 AD 激活启动子从而引发报告基因的转录。
PPT
酵母双杂交系统筛选:用于分离与某一已知蛋白发生相互作用的蛋白质基因
GAL4 蛋白:酵母半乳糖苷酶基因 gal 的转录激活因子,该蛋白结合在 gal 基因上游激活区 (UAS) 可启动 gal 基因的转录
GAL4 蛋白可分为两个区域:
- DNA-BD:DNA 结合域,N-末端 1 ~ 147 aa(氨基酸)
- AD:转录激活域,C-末端 768 ~ 881 aa
这两个结合域将它们分开时仍分别具有功能,但不能激活转录,只有当被分开的两者通过适当的途径在空间上较为接近时,才能重新呈现完整的转录因子活性,并可激活上游激活序列(upstream activating sequence, UAS)的下游启动子,使启动子下游基因得到转录。
酵母双杂交系统筛选原理:
- 将编码 DNA-BD 的基因与已知蛋白质 Bait protein 的基因构建在同一个表达载体上,在酵母中表达两者的融合蛋白 BD-Bait protein。
- 将编码 AD 的基因和 cDNA 文库的基因构建在 AD-LIBRARY 表达载体上。
- 同时将上述两种载体转化改造后的酵母,如两种载体所表达的融合蛋白不相互作用时,这种改造后的酵母细胞的基因组中既不能产生 GAL4,又不能合成 LEU、TRP、HIS、LacZ,因此,酵母在缺乏这些营养的培养基上无法正常生长。当上述两种载体所表达的融合蛋白能够相互作用时,功能重建的反式作用因子能够激活酵母基因组中的报告基因 HIS、LacZ 等,从而通过功能互补和显色反应筛选到阳性菌落。
《分子生物学》P221
酵母双杂交系统巧妙地利用真核生物转录调控因子的组件式结构特征,因为这些蛋白质往往由两个或两个以上相互独立的结构域构成其中 DNA 结合结构域(binding domain,BD)和转录激活结构域(AD)是转录激活因子发挥功能所必需的。
单独的 BD 能与特定基因的启动区结合,但不能激活基因的转录,而由不同转录调控因子的 BD 和 AD 所形成的杂合蛋白却能行使激活转录的功能。
实验中,首先运用基因重组技术把编码已知蛋白的 DNA 序列连接到带有酵母转录调控因子(常为 GAL1、GAL4 或 GCN1)BD 结构域基因片段的表达载体上。导入酵母细胞中使之表达带有 DNA 结合结构域的杂合蛋白,与报告基因上游的启动调控区相结合,准备作为“诱饵”捕获与已知蛋白相互作用的基因产物。此时,若将已知的编码转录激活结构域 AD 的基因片段分别与待筛选的 cDNA 文库中不同插入片段相连接,获得“猎物”载体,转化含有“诱饵”的酵母细胞,只要酵母细胞中表达的“诱饵”蛋白与“猎物”载体中表达的某个蛋白质发生相互作用,不同转录调控因子的 AD 和 BD 结构域就会被牵引靠拢,激活报告基因表达。分离有报告基因活性的酵母细胞,得到所需要的“猎物”载体,就能得到与已知蛋白质相互作用的新基因。
酵母双杂交技术的应用:
- 发现新蛋白质及蛋白质的新功能
- 建立基因组蛋白质连锁图谱
- 研究人类 DNA 文库,筛选药物作用位点
- 在细胞体内研究抗原和抗体的相互作用
参考文献:
- 王婷,葛怀娜,郭宏.酵母双杂交技术应用进展[J].生物技术进展,2015,5(05):392-396.
- 郑立双,李向楠,孙城涛,刘红羽,刘勋,贺明,吕文发.酵母双杂交技术及应用的研究进展[J].中国畜牧兽医,2013,40(09):105-108.
动物基因工程的概念 ↩
动物基因工程是利用 DNA 重组技术对动物所进行的工程操作。从遗传学角度分为遗传性和非遗传性两种形式。
- 外源基因能够通过配子进行垂直传递并稳定遗传的称为遗传性动物基因工程。
- 转基因仅在当代表现,不能够遗传给子代的被称为非遗传性动物基因工程。↩
转基因动物的制备方法主要有哪些? ↩
目前,研究制备转基因动物的主要方法有:
- 显微注射法
- 胚胎干细胞法
- 反转录病毒感染法
- 精子载体导入法
- 体细胞核移植法
简述 CRISPR/Cas9 基因定向打靶技术的原理 ↩
PPT
CRISPR/Cas 系统概述
CRISPR/Cas 系统是很多细菌和大部分古细菌的天然免疫系统,通过对入侵的病毒和核酸进行特异性的识别,利用 Cas 蛋白进行切割,从而达到对自身的免疫。
CRISPR 结构:CRISPR 是一种特殊的 DNA 重复序列家族,广泛分布于细菌和古细菌基因组中。CRISPR 位点通常由短的高度保守的重复序列(repeats)组成,重复序列的长度通常为 21 ~ 48 bp,重复序列之间被 26 ~ 72 bp 间隔序列(spacer)隔开。CIRSPR 通过这些间隔序列(spacer)与靶基因进行识别。
Cas 家族:Cas(CRISPR associated)存在于 CRISPR 位点附近,是一种双链 DNA 核酸酶,能在 guide RNA 引导下对靶位点进行切割。它与 fokⅠ 酶功能类似,但是它并不需要形成二聚体才能发挥作用。
CRISPR/Cas9 系统作用机制
- CRISPR 基因座的表达(包括转录和转录后的成熟加工)
- 当该噬菌体再次入侵细菌时,CRISPR 簇首先转录为长的 crRNA 前体,然后逐步加工成小的成熟的 crRNA。
- CRISPR/Cas 系统活性的发挥或外源遗传物质的干扰
- crRNA 结合相关的 Cas 蛋白后,形成 crRNA-Cas 蛋白复合体,通过碱基互补配对精确地与目标 DNA 相结合,随后 Cas 蛋白对目标 DNA 进行断裂和降解。
- Cas9 是一种核酸内切酶,其具有:RuvC 和 HNH 两个内切酶活性中心
- Jinek 等发现 Cas9 在细菌和试管里对双链 DNA 具有强烈的切割能力,但是这种切割能力需要的 crRNA 和 tracrRNA 介导。
- Jinek 等创造性的把 crRNA 和部分 tracrRNA 融合成一条嵌合的 RNA 链,使其同时具有 crRNA 和 tracrRNA 的特性,随后的切割实验表明,这条嵌合的 RNA 同样可以引导 Cas9 切割目标 DNA。现在普遍使用的都是 crRNA 和全长 tracrRNA 融合体,简称 sgRNA (single guide RNA)
文献资料
该系统原来是细菌及古菌适应性免疫系统的一部分,其功能是抵御病毒及外源 DNA 的入侵。 CRISPR/Cas 系统由 CRISPR 序列和 Cas 基因家族组成。其中,CRISPR 序列由一系列间隔序列及高度保守的正向重复序列相间排列形成,Cas 基因簇位于 CRISPR 序列的 5′ 端,编码的蛋白质可特异性切割外源 DNA。
CRISPR 是一种特殊的 DNA 重复序列家族,广泛分布于细菌和古细菌基因组中。CRISPR 位点通常由短的高度保守的重复序列(repeats)组成,重复序列的长度通常为 21 ~ 48 bp,重复序列之间被 26 ~ 72 bp 间隔序列(spacer)隔开。CRISPR 通过这些间隔序列与靶基因进行识别。
Cas(CRISPR associated)存在于 CRISPR 位点附近,是一种双链 DNA 核酸酶,能在 guide RNA 引导下对靶位点进行切割。它与 fok I 酶功能类似,但是它并不需要形成二聚体才能发挥作用。
CRISPR/Cas 系统分为 I 型、Ⅱ 型和 Ⅲ 型,以 Ⅱ 型的应用最为广泛。有外源 DNA 入侵时,CRISPR 序列转录并被加工形成约 40 nt(核苷酸数) 的成熟 crRNA(CRISPR RNAs)。成熟的 crRNA 与 tracrRNA(trans-activating CRISPR RNA)通过碱基互补配对形成双链 RNA,激活并引导 Cas9 切割外源 DNA 中的原型间隔序列(protospacer)。Cas9 蛋白具有两个核酸酶结构域:RuvC-like 结构域和 HNH 结构域,其中,HNH 结构域切割原型间隔序列中与 crRNA 互补配对的 DNA 链,RuvC-like 结构域切割另一条非互补链。研究表明,Cas9 对靶序列的编辑依赖于原型间隔序列下游的短序列 PAM (protospacer-adjacent motifs),PAM 通常为 5′-NGC-3′,极少情况下为 5′-NAG-3′,Cas9 切割的位点位于 PAM 上游第三个碱基。
根据 CRISPR/Cas9 系统的特点,研究者将 crRNA::tracrRNA 双分子结构融合成具有发夹结构的 sgRNA(single guide RNA),sgRNA 分子 5′ 端 20 nt 的引导序列可完全与 DNA 靶序列互补,从而引导 Cas9 对靶 DNA 进行编辑。只要改变 sgRNA 中的 20 nt 引导序列,基因组上任意 5′-(N)20-NGG-3′ 序列都可以被 CRISPR/Cas9 系统编辑。
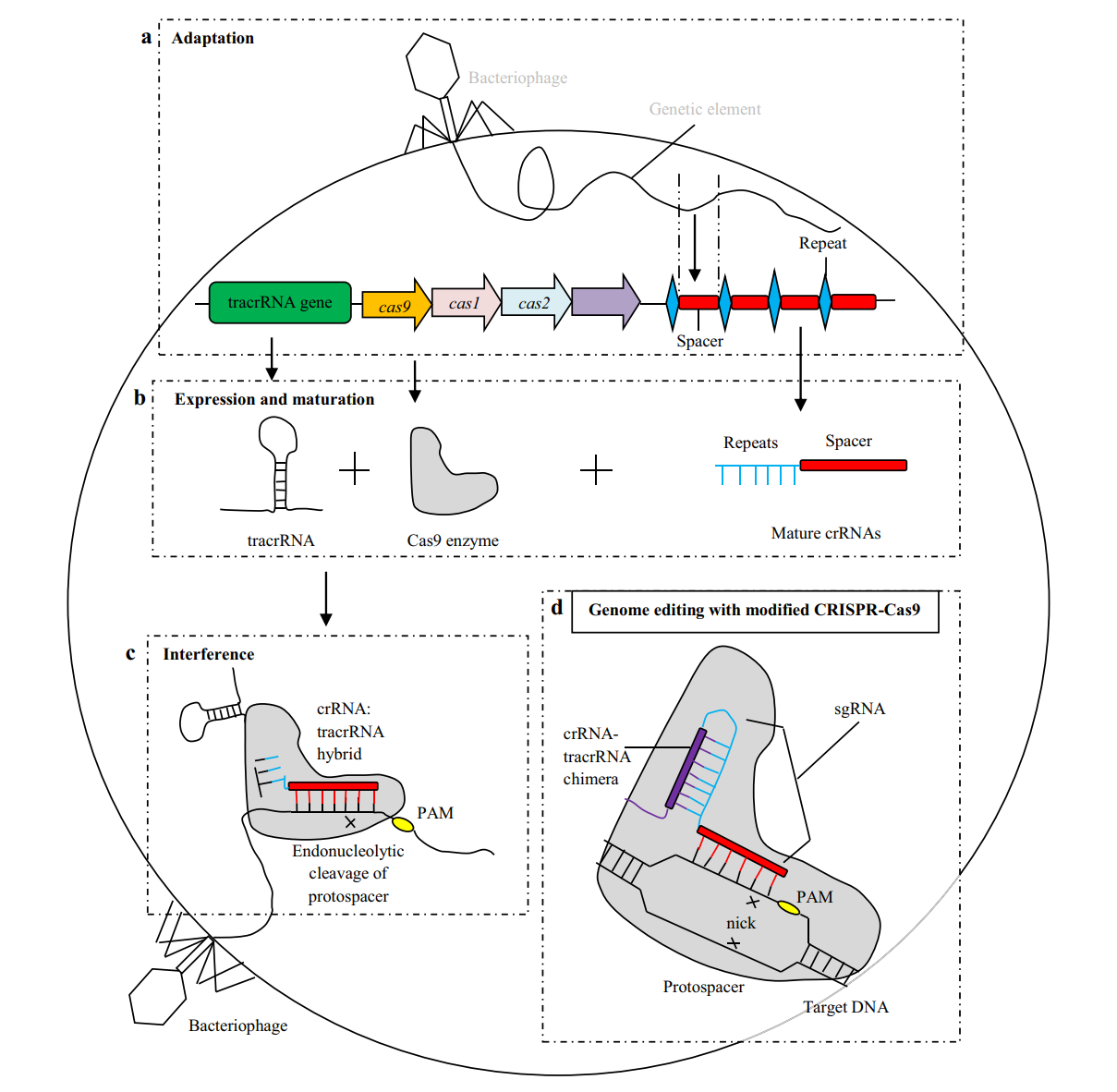
a ~ c 是 CRISPR/Cas9 系统的天然机制,d 是在基因编辑中 crRNA::tracrRNA 双分子结构融合成具有发夹结构的 sgRNA。
引用自 Antara Barman doi:10.1007/s00294-019-01040-3
Lander E S. The heroes of CRISPR[J]. Cell, 2016, 164(1-2): 18-28.
详述将基因打靶技术联合应用胚胎干细胞法制备转基因动物的流程 ↩
- 制备基因打靶载体,把目的基因和调控序列等与内源靶序列都重组到带标记基因的载体上
- 把打靶载体导入胚胎干细胞内,利用点击、脂质体包埋等方法对胚胎干细胞进行转染
- 对同源重组子的筛选,用选择性培养基筛选打靶击中的重组阳性细胞
- 收集囊胚期胚胎,在显微操作系统下向囊胚腔内注入转基因胚胎干细胞,经过短暂培养后,将质量好的囊胚移植到受体子宫内进行妊娠观察
- 筛选所得的转基因动物品系
详述转基因动物技术的应用 ↩
- 研究基因的结构与功能
- 研究基因的组织特异性表达
- 研究发育相关基因的表达与调控
- 克隆在发育中起重要作用的基因
- 基因多级调节系统的研究
- 优良动物品种育种
- 快速生长与肉质改良
- 增强抗病性
- 增强抗逆性
- 研究病毒性疾病
- 研究建立人类疾病的转基因动物模型
- 转基因动物与基因治疗
- 生产天然活性药物蛋白
酵母基因工程的优势有哪些方面? ↩
酵母菌表达外源基因的优势:
- 酵母菌是最简单的真核模式生物
- 完成全基因组测序,基因表达调控机理比较清楚,遗传操作简便
- 具有原核细菌无法比拟的真核蛋白翻译后加工系统
- 大规模发酵历史悠久、技术成熟、工艺简单、成本低廉
- 能将外源基因表达产物分泌至培养基中
- 采用高表达的启动子,如 MOX、AOX、LAC4 等
- 不含有特异性的病毒、不产内毒素,美国 FDA 认定为安全的基因工程受体系统
详述酵母基因工程的应用进展 ↩
利用酵母基因工程成功地生产了人类、动物、植物或微生物来源的异源蛋白,在医药生物技术上发挥了重要作用。例如:
- 生物反应器。人血清白蛋白、溶菌酶、乙肝疫苗等。
- 人类蛋白质间的互作。如利用酵母表达系统开展分泌蛋白和受体基因的快速筛选。
- 筛选新药。利用酵母展示技术筛选细胞凋亡的抑制剂、HIV-1 整合抑制新药等。
- 利用酵母表达膜蛋白。
近年来,酵母基因工程菌已经实现了工业化生产,酵母产生的蛋白已经用在食品、饲料、洗涤、纺织和造纸等行业。尤其在酶制剂领域,国内有近 10 家企业的酶制剂年产量达到万吨,年总产值约 10 亿,并且以每年 10% 左右的速度增长。例如:
- 利用重组酿酒酵母提高酿酒工艺
- 利用重组酿酒酵母环境能源危机。通过转基因技术使酿酒酵母能利用纤维素和半纤维素等可再生物质来生产廉价的酒精,缓解能源紧张。
- 利用毕赤酵母生产饲料用植酸酶。
- 利用酿酒酵母生产谷胱甘肽
什么是基因治疗 ↩
- 经典概念(狭义概念):针对患者的基因缺陷,导入特定基因补偿其缺陷或赋予机体新的功能以抗衡缺陷。
- 广义概念:将目的基因放进特定载体中导入靶细胞或组织,通过替换或补偿引起疾病的基因,调节异常表达的基因来治疗或缓解疾病,均称之为基因治疗。↩
目前基因药物的分类及作用原理 ↩
基因工程药物包括蛋白质类药物和核酸类药物两类:
- 蛋白质类药物主要有重组细胞因子、重组多肽类激素、重组溶栓药物、基因工程抗体和基因工程疫苗等。
- 核酸类药物主要有 DNA 药物,反义核酸药和 RNA 干扰药物等。主要针对的疾病有肿瘤、感染性疾病、人免疫缺陷病毒相关疾病、自身免疫性疾病、器官移植、心血管疾病、神经障碍性疾病等。
基因工程药物的作用机理分为以下三类:
- 蛋白或多肽药物:通过蛋白自身的生理生化特性而抵抗疾病,胰岛素、生长素等激素,各类生长因子。
- 基因工程疫苗、基因工程抗体:基于抗原抗体反应的原理来抵抗疾病;乙肝疫苗,新冠肺炎疫苗,霍乱菌疫苗等。
- 反义核酸、核酶和 RNAi,基于中断基因表达来抵抗疾病。福米韦生,第一代反义核酸药物,治疗艾滋病(AIDS)病人并发的巨细胞病毒(CMV)性视网膜炎。通过对人类巨细胞病毒(CMV) mRNA 的反义抑制发挥特异而强大的抗病毒作用。
详述基因治疗在肿瘤治疗上的主要方向及重大进展 ↩
主要方向有:
- 自杀基因治疗:通过转入自杀基因引起肿瘤细胞表型的改变,从而引起药物对肿瘤细胞直接或间接杀伤作用
- 免疫基因治疗:是把产生抗病毒或者肿瘤免疫力对应的抗原决定族基因导入机体细胞,以达到治疗目的。
- 小分子干扰 RNA 治疗:通过转入外源的小分子 RNA 干扰沉默肿瘤细胞中突变基因的表达
- 增强药物化疗效果:通过外源基因的引入增强药物化疗效果
- 利用基因编辑技术:CRISPR 技术有望进一步改进肿瘤免疫疗法,人工改造的具有嵌合抗原受体的 T 细胞,重新恢复对肿瘤细胞的免疫抑制作用。
仅供参考
参考文献:
- 钟里科.肿瘤免疫治疗的研究进展[J].临床医药文献电子杂志,2017,4(43):8356-8357.
- 孟圆,马晓磊.外源基因治疗肿瘤研究进展[J].济宁医学院学报,2017,40(06):457-461.
- 马云青,常兴.基因编辑与基因治疗[J].生命的化学,2019,39(01):21-27.
基因编辑技术同样在肿瘤治疗领域可能得到有效应用。肿瘤的发生涉及许多基因,包括原癌基因、抑癌基因等。
例如,人子宫颈癌的发生和 HPV (human papilloma virus)感染密切相关。HPV 是 DNA 病毒。HPV 病毒 DNA 在超过 95% 的子宫颈癌患者中被发现。HPV 主要致癌基因为 E6、E7。E6 和 E7 会选择性地表达于 HPV 相关的肿瘤细胞中,使抑癌基因 p53 和 pRb 失活,细胞周期紊乱,端粒酶活化,从而使肿瘤细胞永生。其中 E6 诱导 p53 降解,从而抑制 p53 相关的信号通路,导致癌化。E7 通过与 pRb 蛋白作用使细胞周期调节紊乱。因此,E6 和 E7 基因成为子宫颈癌的基因治疗靶点。目前,体内外实验已证明,CRISPR/Cas9 技术可以靶向 E6 和 E7 编码基因的启动子区域和基因编码区域进行基因敲除,阻止肿瘤细胞增殖。
CRISPR 技术也有望进一步改进肿瘤免疫疗法。肿瘤免疫疗法采取另外一种思路来对抗肿瘤,治疗靶点不再是肿瘤,而是免疫系统。该疗法主要采用具有抗肿瘤功能的单克隆抗体或者供体 T 细胞。肿瘤细胞会发生大量的遗传学改变和表观遗传学修饰,从而产生肿瘤细胞特异性的抗原,正常情况下,宿主免疫系统会识别这些特异性的抗原,并且清除肿瘤。但肿瘤也会进化出相应的免疫逃逸机制。其中一种重要的免疫逃逸机制是免疫抑制信号通路——免疫检查点,正常情况下,免疫检查点会介导免疫耐受,减轻组织损伤;但病理情况下,肿瘤细胞会通过免疫检查点抑制免疫系统的活化逃避免疫系统的监视。其中重要的免疫检查点是细胞毒 T 淋巴细胞相关抗原-4(处 cytotoxic T-lymphocyte-associated antigen-4, CTLA-4)和程序性死亡分子 PD-1(programmed death-1, PD-1)
为了重新恢复对肿瘤细胞的免疫抑制作用,采用人工改造的具有嵌合抗原受体的 T 细胞 [chimeric antigen receptor (CAR) T cells]。嵌合抗原受体在 T 细胞外部具有针对肿瘤细胞特异性抗原的结合结构域,在 T 细胞内部具有 T 细胞激活结构域和 T 细胞肿瘤细胞杀伤功能促进结构域。但是绝大数的 CART 来源于病人自身T细胞,时间成本和费用成本都很高,相对常规的抗体封闭免疫检查点的方法并不具有优势。通用的 CART 细胞会降低成本,但会与受体彼此间产生免疫排斥。ZFN 技术和 TALEN 技术已经实现了 T 细胞中内源性 TCR 基因敲除以及 CART 细胞中主要组织相容性抗原的敲除,从而解决免疫排斥问题。除此之外,为进一步加强 CART 的功能,有学者利用基因编辑技术将 CTLA-4 或 PD-1 基因敲除。
第一章 基因工程概论
基因工程的诞生
- 基因工程:又称 DNA 重组技术,是指以分子遗传学为理论基础,以分子生物学和微生物学的现代方法为手段,将不同来源一种或多种基因按预先设计的蓝图,在体外构建杂种 DNA 分子,然后导入活细胞,使之按照人们的意愿遗传并表达出新的性状,从而创造出人们所需求的新的生物类型。↩
- 基因工程要素:包括外源 DNA;工具酶;载体分子和受体细胞等。
基因工程诞生的理论基础
- 孟德尔提出遗传因子分离定律和自由组合定律
- 摩尔根并提出一条染色体决定一个性状
- 1944年,Avery 利用肺炎双球菌转化实验证明了 DNA 是遗传物质,DNA 可从一种生物个体转移到另一种生物个体。
1944年,美国洛克菲勒研究所的 Oswald Avery 等公开发表了改进的肺炎双球菌实验结果。
- S型菌细胞提取物及其纯化的DNA都可使R型菌转变成S型菌;
- 经 DNase 处理的 S 型菌细胞提取物失去了转化作用。
- 经胰蛋白酶处理的 S 型菌细胞提取物仍有转化作用。
实验证实了DNA是遗传物质,而且证明了DNA可以将一个细菌的性状转给另一个细菌,他的工作被称为是现代生物科学的革命性开端。
- DNA 双螺旋结构,1953 年 James D. Watson 和 Francis H. C. Crick 揭示了 DNA 分子的双螺旋结构和半保留复制机制。解决了基因的自我复制和传递问题
- 中心法则和遗传密码,以 Nireberg 等为代表的一批科学家经过艰苦的努力,确定了遗传信息以密码方式传递,每三个核苷酸组成一个密码子,代表一个氨基酸,到 1966 年,全部破译了 64 个密码子,并提出了遗传信息传递的“中心法则”。1963 年尼伦伯格和马太破译编码氨基酸的遗传密码,1966 年霍拉纳用实验加以证明。
综上:
- DNA 是遗传物质的证明
- DNA 双螺旋结构和中心法则的确立
- 遗传密码的破译(遗传密码的通用性)
基因工程诞生的技术突破
- 基因的剪刀——限制性内切酶
- 基因的针线——DNA连接酶
- 基因的运输工具——运载体
| 工具 | 作用 |
|---|---|
| 限制性内切酶 | 在特定的切点上切割 DNA 分子 |
| DNA连接酶 | 把两条 DNA 末端间的缝隙“缝合”起来 |
| 运载体 | 将外源基因送入受体细胞 |
限制性内切酶:
- 1968 年,Werner Arber 发现限制酶
- 1970 年,H.O. Smith 等分离并纯化了限制性核酸内切酶Hind Ⅱ
- 1972 年,Boyer 等相继发现了EcoR Ⅰ 一类重要的限制性内切酶。
DNA 连接酶:
- 1967 年,世界上有五个实验室几乎同时发现 DNA 连接酶
- 特别是 1970 年 Khorana 等发现的 T4 DNA 连接酶具有更高的连接活性。
DNA连接酶连接形成磷酸二酯键
载体的发现与应用:
- 1972 年前后使用小分子量的细菌质粒和 λ 噬菌体作载体。在细菌细胞里大量扩增。
- 1973 年,Stanford 大学的 Cohen 等成功地利用体外重组实现了细菌间性状的转移。这一年被定为基因工程诞生的元年。
感受态体系:
- 1970 年 M. Mandel 和 A. Higa 发现经过氯化钙处理的大肠杆菌容易吸收噬菌体 DNA。
- 1972 年 S. Cohen 发现这种处理过的细菌同样能吸收质粒 DNA。
琼脂糖凝胶电泳:1960s 发明了琼脂糖凝胶电泳,可将不同长度的 DNA 分离开。
DNA 测序技术:1975 年 F. Sanger、A. Maxam 和 W. Gilbert 发明了 DNA 快速测序技术。
基因工程的支撑技术
- 核酸凝胶电泳技术
- 核酸分子杂交技术
- 细菌转化转染技术
- DNA 序列分析技术
- 寡核苷酸合成技术
- 基因定点突变技术
- 聚合酶链反应(PCR)技术
基因工程的诞生
Berg 的开创性实验,1972 年斯坦福大学的 Paul Berg 小组完成了首次体外重组实验:用EcoR Ⅰ 内切酶消化猿猴病毒 SV40 的 DNA 和 λ 噬菌体的 DNA,再用 T4 DNA 连接酶将其连接起来。
Boyer-Cohen 实验,1973 年斯坦福大学 S. Cohen 小组将含有新霉素抗性基因的大肠杆菌 R6-3 质粒与含有四环素抗性基因的质粒 pSC101 连接成重组质粒,转化获得具有双重抗药性的大肠杆菌。后来又把非洲爪蟾核糖体基因片断同 pSC101 质粒重组,转化大肠杆菌,并在菌体内成功转录出相应的 mRNA。这是第一次成功的基因克隆实验。
此实验说明:
- PSC101 可以做为载体,将外源 DNA 导入寄主细胞
- 真核生物的基因可以转录到原核生物
- 质粒-大肠杆菌是成功的基因克隆体系
基因工程的特征
- 跨物种性:将一外源基因转到另一种不同的生物细胞内进行繁殖。
- 无性扩增:外源 DNA 在寄主细胞内可大量扩增和表达。
基因工程的研究内容
| 基因工程的别名 | 基因拼接技术或 DNA 重组技术 |
|---|---|
| 操作环境 | 生物体外 |
| 操作对象 | 基因 |
| 操作水平 | DNA 分子水平 |
| 基本过程 | 剪切 → 拼接 → 导入 → 表达 |
| 结果 | 人类需要的基因产物 |
基因工程的基本流程
- 目的基因的获取,从复杂的生物基因组中,经过酶切消化或 PCR 扩增等步骤,分离出带有目的基因的 DNA 片断。(切)
- 重组体的制备,将目的基因的 DNA 片断插入到能自我复制并带有选择性标记(抗菌素抗性)的载体分子上。(接)
- 重组体的转化,将重组体(载体)转入适当的受体细胞中。(转)
- 重组子的培养,对转入重组子的受体细胞进行培养,以扩增 DNA 重组子或使其整合到受体细胞的基因组中。(增)
- 克隆的鉴定,挑选转化成功的细胞克隆(含有目的基因)。(检)
- 目的基因的表达,使导入寄主细胞的目的基因表达出我们所需要的基因产物。
基因工程的应用
基因工程在农业生产中的应用
- 提高植物的光合作用效率:
- 提高 CO2 的固定率,利用蓝藻基因,改变与光合作用有关的酶的结构和组成(如二磷酸核酮糖羧化酶)。
- 提高光能吸收率和转化率,改变光能交换系统的分子的基因结构。
- 提高豆科植物的固氮效率,使非固氮植物转变为固氮植物或能与根瘤菌共生固氮。
- 转基因植物,是农业生物技术的主要内容。将克隆到的特殊基因导入受体植物,使之增加一些优质性状(高产、稳定、优质、抗虫、抗病等)。例如:延熟(耐贮存)番茄、高产量的番茄、抗环斑病毒的转基因番木瓜等
- 转基因动物,将外源基因导入动物细胞,并在基因组内稳定整合,遗传给后代。如使动物成为生物反应器生产有用的活性蛋白等。
基因工程在工业中的应用
- 纤维素的开发利用,克隆各种参与纤维素降解的酶的基因,导入酿酒酵母,就可能利用廉价的纤维素来生产葡萄糖,发酵成酒。
- 酿酒工业,用外源基因改造酿酒酵母,产生优质的啤酒。或用酿酒酵母生产蛋白质等。
基因工程在医药上的应用
- 用转基因植物或动物生产药物
- 用微生物生产药物,大肠杆菌或酵母菌生产激素(如胰岛素)、干扰素等
- 设计高效特异的生物制剂,应用定点突变技术设计蛋白质或酶的结构,制造出高效高特异性的生物制剂
- 研制疫苗,制造新型疫苗(如HIV、癌症、乙肝、丙肝等)
- 基因诊断
- 法医鉴定
- 基因治疗,将正常的外源基因导入靶细胞中以弥补靶细胞所缺失或突变的基因、或抑制异常表达的基因。(仍在探索阶段)
基因工程在环境保护中的应用
- 检测水污染,用重组细菌或转基因鱼等检测水污染
- 生物降解,用带有重组质粒的“超级菌”分解油(烷烃类)、有机农药污染。
基因工程的商业化发展
商业投资支持现代生物技术研究,生物技术的发展在某种程度上是由商品经济的发展所推动的。
基因工程商业化特点:
- 技术密集型
- 产品来源于实验室
- 科学家往往就是公司的领导人
- 市场扩张迅速
- 投入巨大
- 风险太高
- 产品不断增加
- 研究专一、产品专一
- 医学生物技术产业进展最快
第二章 基因操作的工具酶
| 工具酶 | 功能 |
|---|---|
| 限制性内切酶 | 主要用于 DNA 分子的特异切割 |
| DNA 甲基化酶 | 用于 DNA 分子的甲基化 |
| 核酸酶 | 用于 DNA 和 RNA 的非特异性切割 |
| 核酸连接酶 | 用于 DNA 和 RNA 的连接 |
| 核酸聚合酶 | 用于 DNA 和 RNA 的合成 |
| 核酸末端修饰酶 | 用于 DNA 和 RNA 的末端修饰 |
| 其它酶类 | 用于生物细胞的破壁,转化,核酸纯化,检测等。 |
限制性核酸内切酶
细菌的限制与修饰系统
任何物种都有排除异物,保护自身的防御机制
- 人的免疫系统(immunity system)
- 细菌的限制与修饰系统(restriction and modification system):限制性内切酶(restriction endonuclease)、甲基化修饰酶 (modification enzyme)
细菌的限制系统,限制(Restriction)侵入细菌体内的外源 DNA(非甲基化),能被限制性内切酶识别和降解,从而保护自身的 DNA 不被降解。
细菌的修饰系统,细菌自身的 DNA 可被甲基化酶修饰,从而防止限制性内切酶的识别和水解。
Dam 甲基化酶 在 GAATTC 序列的腺嘌呤 N6 位引入甲基。
限制与修饰作用:由于甲基化酶具有种属专一性,即只修饰寄主本身的 DNA,因而避免了限制酶对寄主 DNA 的破坏;对于外源 DNA,甲基化酶不能识别,而由限制酶将其水解。
R-M 系统是细菌安内御外的积极措施。
- 限制:细菌为防御外来 DNA 入侵而将其降解的现象。(一般由限制酶来降解外源 DNA)
- 修饰:细菌为防止自身 DNA 被降解而修饰自身 DNA。(一般由甲基化酶进行修饰)
个别噬菌体在被降解之前已经发生了修饰,则可免予被降解。
限制与修饰系统的意义:
- 是许多细菌所具有的类似免疫的防卫机制。
- 是病毒具有一定寄主范围,感染具有种属专一性的原因。
- 既保证物种的遗传稳定性,又利于生物进化。
限制性内切酶的发现
限制性核酸内切酶(restriction endonuelease):是一类能识别双链 DNA 中特定核苷酸序列,并使每条链的一个磷酸二酯键断开的内脱氧核糖核酸酶(endo-deoxyribonuclease)。
- 1968 年,Werner Arber 发现限制酶
- 1968 年,Meselson and Yuan 从 E. coli 菌株 K 和 B 中发现了Ⅰ型限制酶;
- 1970 年,Smith 等人首先从流感嗜血杆菌 d 株中分离出 Hind Ⅱ和 Hind Ⅲ
限制性内切酶的命名
限制酶由三部分构成,即菌种名、菌株号、分离顺序。
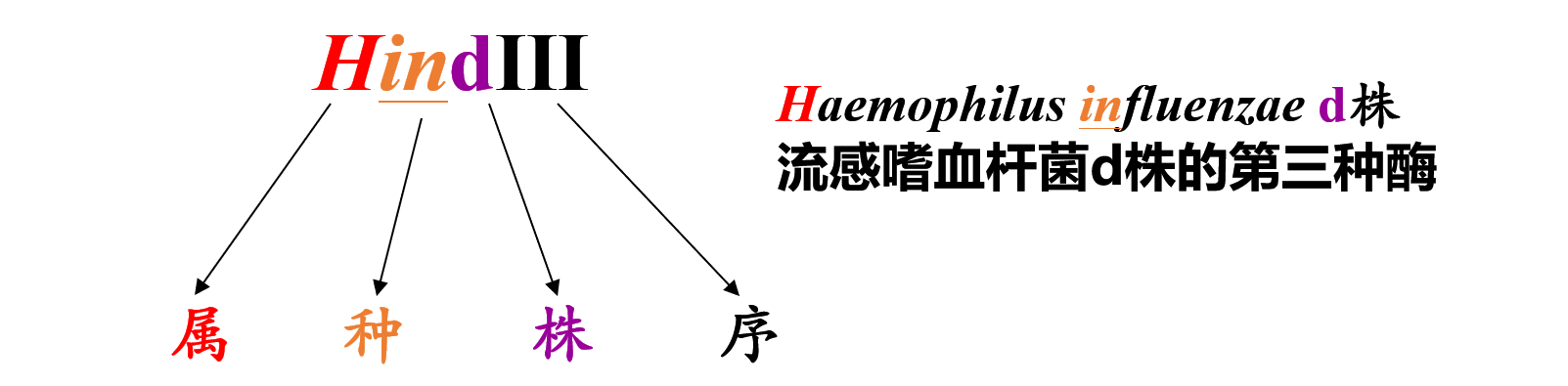
- 第 1 个字母取自产生该酶的细菌属名,大写
- 第 2,3 二个字母取自它来源细菌的种名的头 2 个字母,斜体小写
- 第 4 个字母代表菌株号,大小写都可以,可有可无
- 最后用大写罗马数字,代表同一菌株中不同限制酶的编号
EcoR Ⅰ、HindⅢ、Acc65 Ⅰ、Pst Ⅰ
限制性内切酶的分类
根据限制性内切酶的识别位点和切割位点以及所需要的辅助因子,可将目前已鉴定出的限制性内切酶一般将限制酶分为 3 种不同类型。
- Ⅰ 型:切割位点不确定,不适用于基因工程。
- Ⅱ 型:基因工程的工具酶。
- Ⅲ型:切割位点不在识别位点,对分子克隆操作亦无实用意义。
各种限制与修饰系统的比较
| Ⅰ | Ⅱ | Ⅲ | |
|---|---|---|---|
| 酶分子 | 三亚基多功能酶 | 内切酶与甲基化酶分子不在一起 | 二亚基双功能酶 |
| 识别位点 | 二分非对称 | 4 ~ 6 bp,大多数为回文对称结构 | 5 ~ 7 bp 非对称 |
| 切割位点 | 无特异性,至少在识别位点外 1000 bp | 在识别位点中或靠近识别位点 | 在识别位点下游 24 ~ 26 bp |
| 识别未甲基化的序列进行切割 | 能 | 能 | 能 |
| 限制作用是否需用 ATP | 是 | 否 | 是 |
Ⅱ 型限制性内切酶
首先由 H.O. Smith 和 K.W. Wilcox 在 1970 年从流感嗜血杆菌 Rd 菌株中分离出来。分离的第一个酶是 Hind Ⅱ
作用原理:识别双链 DNA 上未甲基化修饰的一小段明确的序列(多数是回文序列),然后在识别位点之内的特定位置切割。
Ⅱ 型限制性内切酶的特性:一般识别和切割 4 和 6 个核苷酸顺序,少数为 5、7 个核苷酸,也有识别 8 个核苷酸的,但无 4 个以下的。
限制酶切割的位置
切割的位点:
- 一般在识别序列内部,如 G↓GATCC、AT↓CGAT、GTC↓GAC、CCGC↓GG、AGCGC↓T 等。
- 少数在识别序列的两侧,如 ↓GATC、CATG↓、↓CCAGG 等
限制酶切后产生两个末端,末端结构是 5’-P 和 3’-OH
限制酶产生的末端
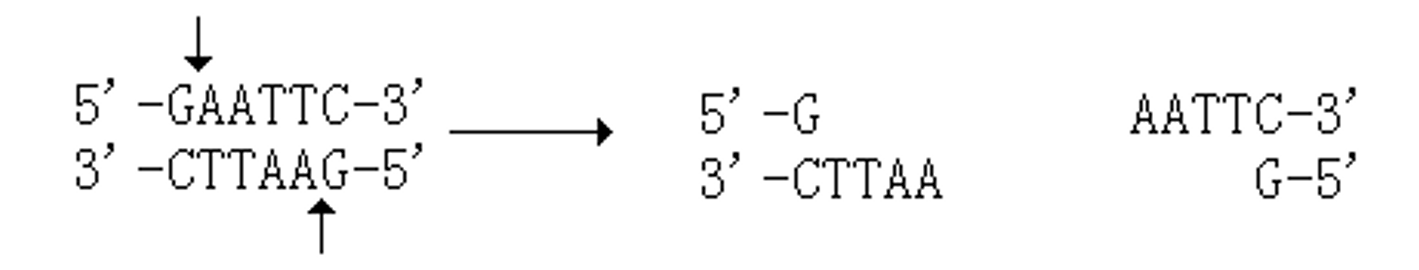
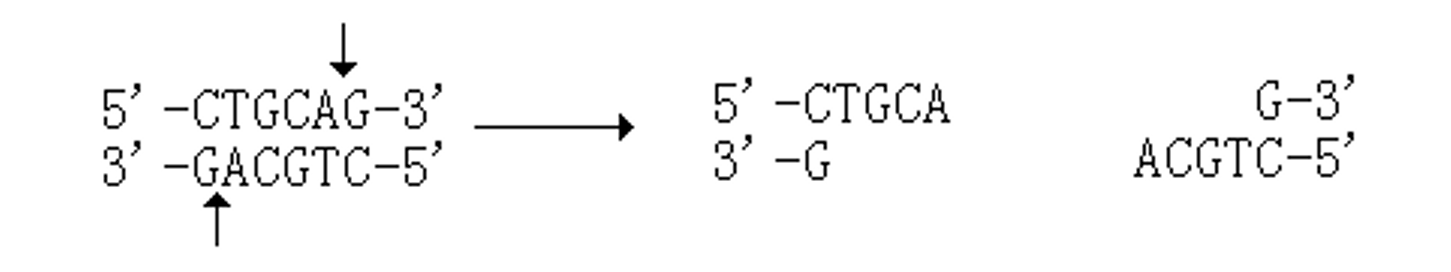
粘性末端:在识别序列 2 条链对应位上错位切割,有 5’ 端突出的粘性末端和 3’ 端突出的粘性末端。 ↩
齐平末端:在识别序列内的对称轴上切割,其切割产物具平头末端(可任意连接,但连接效率低)。
粘性末端的意义:粘性末端突出的单链部分可以与相同的酶或同尾酶切割得到的粘性末端的单链部分互补配对。
- 在回文对称轴上同时切割 DNA 的两条链,则产生平末端
- 若在对称轴 5’ 侧切割底物, DNA 双链交错断开产生 5’ 突出粘性末端
- 若在对称轴 3’ 侧切割底物,则产生 3’ 突出粘性末端
关于限制酶的几个概念

限制酶识别序列同 DNA 的来源无关,也就是说不具有种的特异性,对不同的 DNA 普遍适用。原则上任何不同来源的 DNA,经过适当限制酶消化成片段后,都可以通过其黏性末端或平末端连接起来,组合成新的重组分子或是新的基因。这种特性是重组 DNA 技术的重要基础之一。
同裂酶:识别序列相同而来源不同的一类限制酶。亦称异源同工酶。它们的切割位点可能不同。↩
同裂酶应用:一些同裂酶间对于切割位点上的甲基化碱基的敏感度有所不同,利用这样细微的差异可以进行 DNA 甲基化作用的研究。
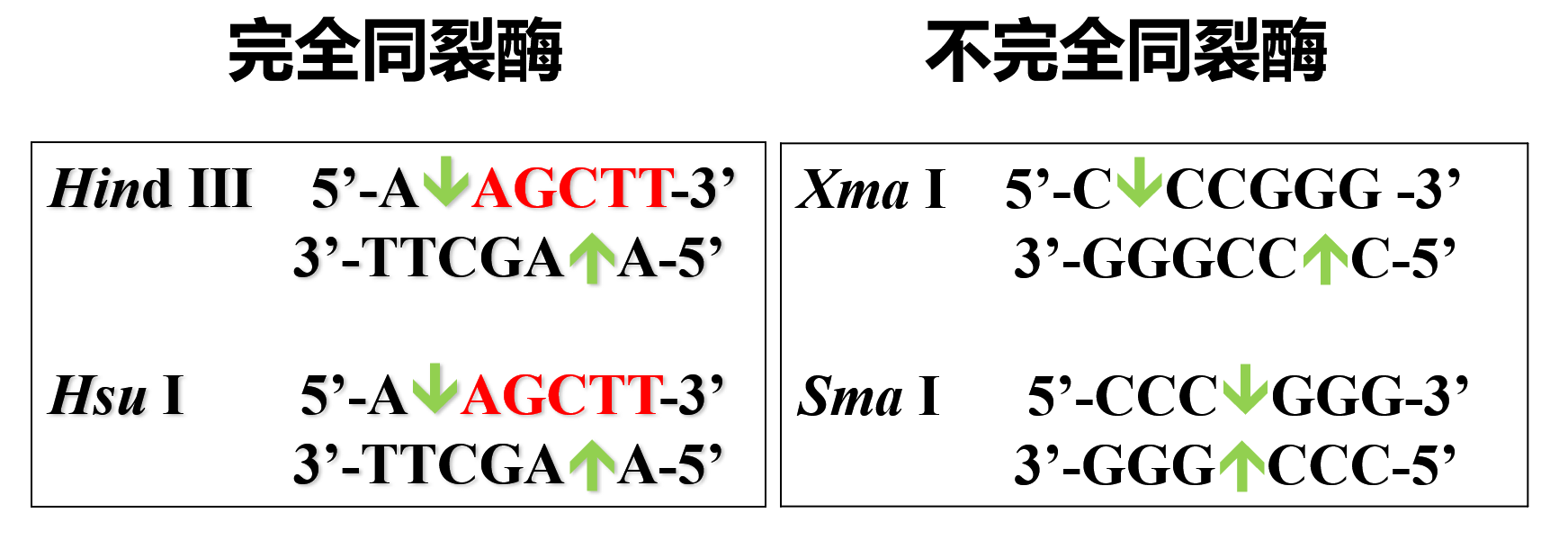
同尾酶:这一类的限制酶来源各异,识别的靶序列也不相同,但产生相同的粘性末端。↩
同尾酶的粘性末端结合形成的新位点不能再被原来的酶识别
酶的星号活性(星星活性):当酶切条件改变时,酶的专一性可能会降低,以至于同一种酶可识别和切割多个的位点。详见 星星活性
双酶切方法:同步双酶切、分步双酶切
Ⅱ 型核酸内切酶的多酶联合酶解,对盐浓度要求不同的酶,可采取下列方法:
- 使用较贵的酶的盐浓度,加大便宜酶的用量,同时酶解
- 低盐酶先切,然后补加盐,高盐酶再切
- 一种酶先切,然后更换缓冲液,另一种酶再切
Ⅲ 类限制性内切酶,能完全肯定的识别位点和切割位点,但切割位点也是在识别位点的一侧的一定距离,通常距特异性位点 24 bp ~ 26 bp。在基因工程操作中用途不大
常用限制性核酸内切酶的特性

限制性内切酶的活性单位
20 μL 中,含 1 μg 底物 DNA,于最适反应条件和温度下,保温 1 小时,能使 1 μg DNA 完全降解所需的酶蛋白量即为一个酶单位,用 U 表示。
限制性内切酶的反应条件
20 μL 的酶切反应体系:
| 总体积 | 20 μL |
|---|---|
| DNA | 1 μL(1 μg) |
| buffer(10x) | 2 μL |
| ddH2O | 16 μL |
| 限制性内切酶 | 1 μL |
- 确定酶切 DNA 的量
- 计算完全酶切所需的酶量。
- 10X 反应液的体积应为终体积的 1/10
- 依次加入计算好的 DNA 量、10X 反应液、酶。
影响限制性内切酶活性的因素
- DNA 的纯度,DNA 中的杂质如蛋白质、酚、氯仿、乙醇、SDS、EDTA 等都会影响酶的活性。一般采取以下措施:纯化 DNA、加大酶的用量、延长酶催化反应的保温时间、扩大反应体积(>20 μl)
- DNA 的甲基化程度,大肠杆菌一般有两种甲基化酶修饰质粒:dam 甲基化酶(修饰 GATC 中的 A);dcm 甲基化酶(修饰 CCA/TGG 的 C)。基因工程中必须使用甲基化酶失活突变的菌株。
- 温度,不同的限制性内切酶的最适反应温度不同。大多数是 37℃,少数要求 40 ~ 65℃。
- DNA 的分子结构,DNA 分子的不同构型对限制性内切酶的活性也有很大的影响。某些限制性核酸内切酶切割超螺旋的质粒 DNA 所需要的酶量要比消化线性的 DNA 量高出很多倍。
- 缓冲液,是影响限制酶活性的重要因素。商品化的限制酶一般都带有专用缓冲液。化学组成:
- MgCl2、NaCl/KCl:提供 Mg2+ 和离子强度
- Tris-HCl:维持 pH
- 二硫苏糖醇(DTT):保持酶中 -SH 的还原态,保持酶稳定性
- 牛血清白蛋白 BSA 等:有助于酶的稳定
限制性内切酶的应用
- 重组 DNA 前的切割
- 构建新质粒
- 构建物理图谱
- DNA 分子杂交
- 用限制性内切酶消化受体 DNA
- 制备 DNA 探针
- 亚克隆以用作序列分析
- 基因定位,DNA 同源性研究
DNA 连接酶
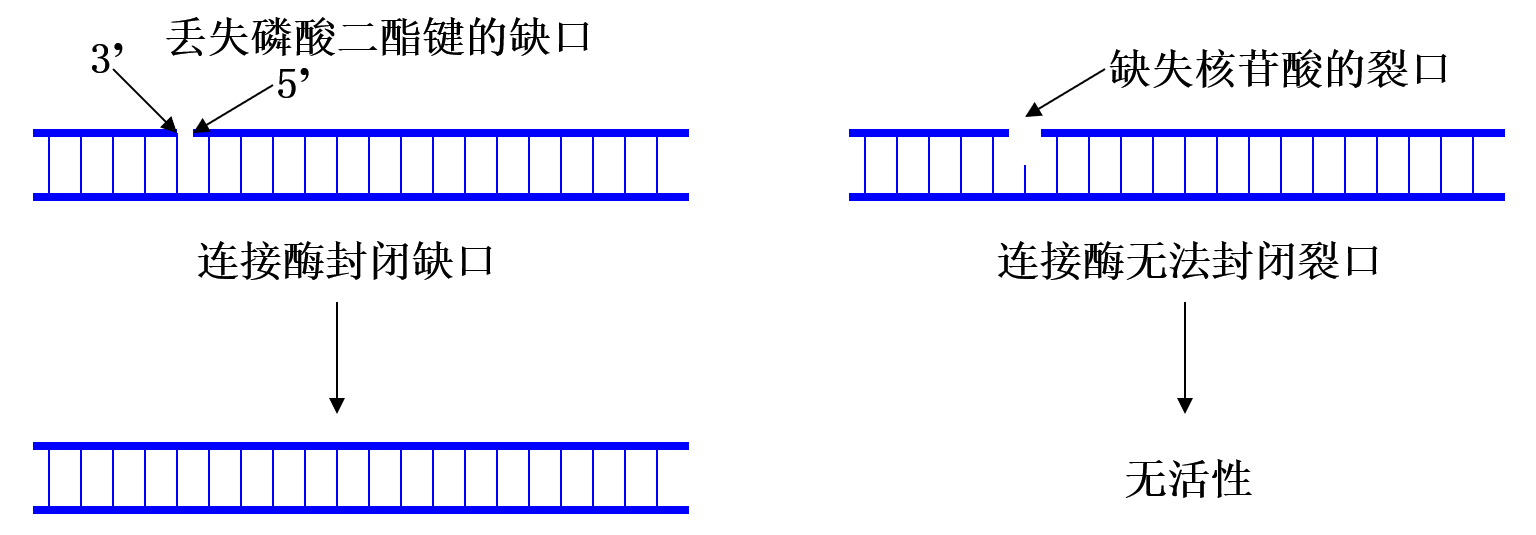
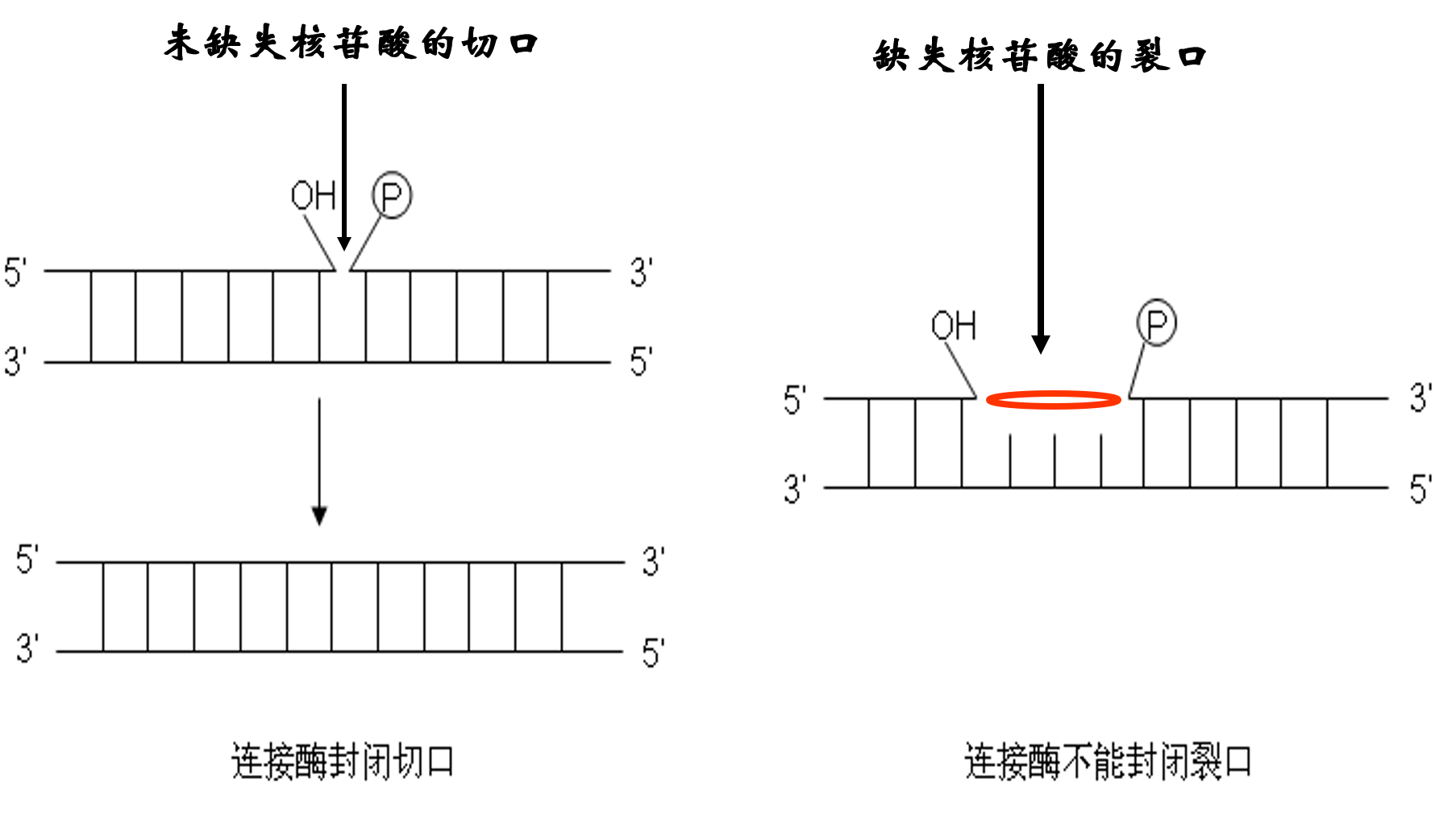
DNA 连接酶:能够催化双链分子中相邻的 3’-OH 和 5’-P 末端之间形成磷酸二酯键,使 DNA 分子连接起来的酶。 ↩
DNA 连接酶种类
- T4 DNA 连接酶,从 T4 噬菌体感染的大肠杆菌提取,T4 DNA ligase 能量 ATP
- 大肠杆菌 DNA 连接酶,E. coli DNA ligase 能量 NAD﹢
DNA 连接酶的连接范围:
- 粘性末端 DNA 连接
- 缺口的填补
- 平末端 DNA 连接

E. coli DNA连接酶或 T4 DNA 连接酶可把黏性末端之间的缝隙“缝合”起来,恢复被限制酶切开的两个核苷酸之间的磷酸二酯键,T4 DNA连接酶还可把平末端之间的缝隙“缝合”起来,但效率较低
DNA 连接酶的连接条件
- 双链 DNA 分子
- 具有 3’-OH 和 5’-P
- 缺口处不缺核苷酸
- 需要能量
T4 DNA 连接酶的反应条件
| 总体积 | 20 μL |
|---|---|
| 10× T4 DNA Ligase Buffer | 2 μL |
| DNA 片段 | 约 0.3 pmol |
| 载体 DNA | 约 0.03 pmol |
| T4 DNA Ligase | 1 μL |
| ddH2O | up to 20 μL |
反应温度:16 ℃,连接过夜
DNA 片段的摩尔数应控制在载体 DNA 摩尔数的 3 ~ 10 倍。
体外 DNA 片段的连接方法
由于待连接的 DNA 片段具不同形式的末端,因此须采用不同的连接方法。
- 具互补黏性末端片段之间的连接
- 具平末端 DNA 片段之间的连接
- DNA 片段末端修饰后进行连接
平头双链DNA片段的连接操作。从分子动力学的角度讲,由限制性核酸内切酶创造的粘性末端的连接属于分子内部的连接,而平头末端的连接则属于分子间的连接,因此后者反应速度要慢得多,提高平头末端连接效率的方法包括:
- 加大连接酶用量(10 倍大于粘性末端的连接)
- 加大平头末端底物的浓度,增加分子间碰撞机会
- 加入10% PEG8000,促进大分子之间的有效作用
- 加入单价阳离子(NaCl),最终浓度 150 ~ 200 mM
平头双链 DNA 片段修饰后进行连接:
- DNA 片段末端同聚物加尾后进行连接。给平末端 DNA 片段 3’-OH 加上同聚物。
- DNA 片段加连杆或衔接头后连接
DNA 接头(adapter)连接法:于 1978 年由康乃尔大学吴瑞教授发明的。它是一类由人工合成的一头具有某种限制性内切酶粘末端,另一头为平末端的特殊的双链寡核苷酸片段。当它的平末端与平末端的外源 DNA 片段连接之后,便会使后者成为具黏性末端的新的 DNA 分子,而易于连接重组。
小结:平末端 DNA 片段的连接
- 直接用 T4 DNA 连接酶连接;
- 先用末端核苷酸转移酶,给平末端 DNA 分子加上同聚物尾巴之后再用 DNA 连接酶进行连接;
- DNA 接头连接法。
DNA 聚合酶
常用的 DNA 聚合酶
- 大肠杆菌 DNA 聚合酶 Ⅰ
- T7 DNA聚合酶
- Taq DNA聚合酶
- Pfu DNA聚合酶
- 末端转移酶
- 反转录酶
DNA 聚合酶的特点
- 共同特点,在 DNA 模板指导下,以 4 种脱氧核糖核苷酸(dNTP)为底物,在引物 3’-OH 末端聚合 DNA 链(DNA合成)的一类酶。
- 主要区别:
- 持续合成能力和外切酶活性不同。
- T7 DNA 聚合酶可以连续添加数千个 dNTPs 而不从模板上掉下来。
- 其它几种 DNA 聚合酶只能连续添加 10 多个 dNTPs 就会从模板上解离下来。
DNA 聚合酶的特性比较:
| DNA 聚合酶 | 3’→5’ 外切酶活性 | 5’→3’ 外切酶活性 | 聚合速率 | 持续能力 |
|---|---|---|---|---|
| 大肠杆菌DNA聚合酶 | 低 | 有 | 中 | 低 |
| Klenow fragment | 低 | 无 | 中 | 低 |
| T4 DNA聚合酶 | 高 | 无 | 中 | 低 |
| T7 DNA聚合酶 | 高 | 无 | 快 | 高 |
| 逆转录酶 | 无 | 无 | 低 | 中 |
| Taq DNA聚合酶 | 无 | 有 | 快 | 高 |
DNA聚合酶的活性:
- 5’→3’ 聚合酶活性
- 5’→3’ 外切酶活性
- 3’→5’ 外切酶活性
大肠杆菌 DNA 聚合酶Ⅰ
功能:
- 5’→3’ 聚合酶活性
- 5’→3’ 外切酶活性
- 3’→5’ 外切酶活性
聚合反应的条件:
- 底物为四种脱氧核苷 5’ 三磷酸(dNTPs)。
- 引物为 3’ 端具有游离的 -OH 基团。
- 模板为单链 DNA 或具有切口的双链 DNA。
主要用途:通过 5’→3’ 聚合酶活性和 5’→3’ 外切酶活性使 DNA 切口平移,制备核酸分子杂交用的带放射性标记的 DNA 探针。
制备 DNA 分子杂交探针(缺口平移 Nick translation)
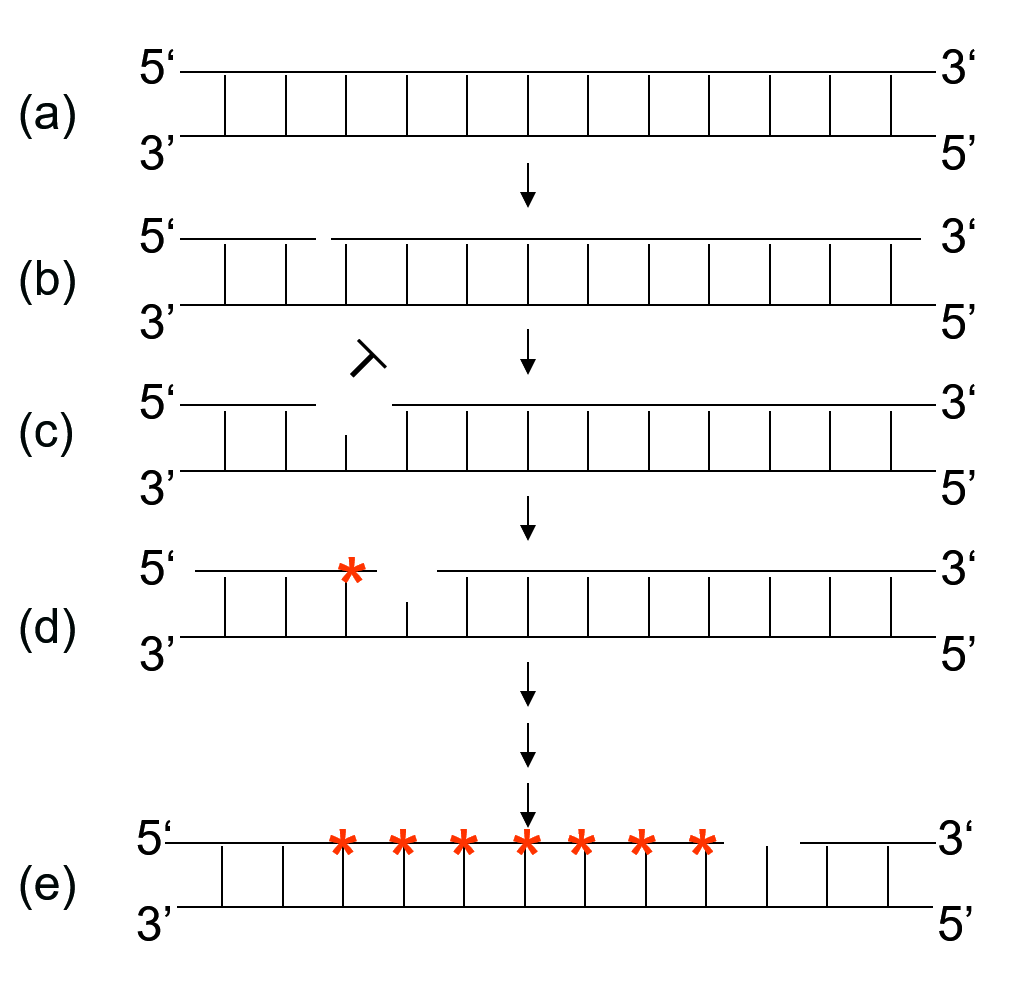
- DNase Ⅰ 处理双链的 DNA 分子
- 带有 3’-OH 末端的单链缺口
- polⅠ从 5’-P 移去一个核苷酸
- polⅠ将 32P 标记的核苷酸参入取代被移去的核苷酸
- 重复 3、4 的步骤,缺口沿 5’-3’ 方向移动,形成 32P 标记的核苷酸合成的 DNA 链
核酸探针(probe):能够同某种被研究的核酸序列特异性结合的、带有标记的寡聚核苷酸。
- 标记:已知序列的核酸片段
- 显示位置:与互补的待测序列杂交
T7 DNA 聚合酶的特点及用途
- 持续合成能力强,一旦与模板结合就会不间断地合成互补链。
- 3’→ 5’ 外切酶活性高,单链和双链都能降解。
- 不受 DNA 二级结构的影响,其它 DNA 聚合酶受 DNA 二级结构的阻碍
用途:用于测序
Taq DNA 聚合酶
由嗜热的水生菌 Thermus aquaticus 分离纯化,主要用于 DNA 测序、聚合酶链式反应(PCR)体外扩增 DNA。
因为没有 3’→5’ 外切酶活性,所以有一定的错配率,约 1000 bp 有 2 ~ 3 bp 错配。
Pfu DNA 聚合酶
Pfu 聚合酶有着出色的热稳定性,以及特有的“校正作用”。与 Taq DNA 聚合酶不同,Pfu DNA 聚合酶具有 3’-5’ 外切酶的即时校正活性,可以即时的识别并切除错配核苷酸。
商业化的 Pfu 聚合酶试剂,其出错率是 100 万到 130 万个碱基对出现一个错配,因此,Pfu 聚合酶正逐渐取代 Taq 聚合酶,成为使用最广的 PCR 工具。
末端转移酶
末端转移酶(terminal transferase),(不依赖模板的 DNA 聚合酶)
用途:
- 克隆 DNA 片段时加上互补同聚物末端,便于与载体连接。
- 末端标记
末端转移酶催化 DNA 片段在其 3′ 羟基末端加接脱氧核糖核苷酸。合成时不需 DNA 模板,但是底物要有一定长度,至少是 3 个碱基以上的寡核苷酸片段。反应底物可以是带有 3′ 羟基的单链 DNA,也可以是 3′ 端延伸的双链 DNA,反应时 4 种 dNTP 的任何一种都可以作为合成的前体物。
TdT 的基本特性:不需要模板的 DNA 聚合酶,随机掺入 dNTPs。
反转录酶:AMV
来源,商品反转录酶有两种:
- 来自禽类成髓细胞瘤病毒(AMV)
- 来自 Moloney 鼠白血病毒(M-MLV)反转录酶
结构和活性:
| 酶类别 | 肽链 | 5’-3’ 聚合活性 | RNase H |
|---|---|---|---|
| AMV | α(62 KDa) 、 β(94 KDa) | + | +++ |
| M-MLV | 84 KDa | + | + |
α 链:有反转录活性和 RNaseH 活性
RNaseH:α 链经过蛋白酶水解后产生的一条多肽。以 5’→3’ 或 3’→5’ 方向特异地水解 RNA-DNA 杂交双链中的 RNA 链。
β 链:RNA-DNA 杂交双链中 5’→3’ DNA 外切酶活性
DNA 连接酶与 DNA 聚合酶的异同
| DNA聚合酶 | DNA连接酶 | |
|---|---|---|
| 区别1 | 只能将单个核苷酸连接到已有的核酸片段上,形成磷酸二酯键 | 在两个DNA片段之间形成磷酸二酯键 |
| 区别2 | 以一条DNA链为模板,将单个核苷酸通过磷酸二酯键连接成一条互补的DNA链 | 将DNA双链上的两个切口同时连接起来,不需要模板 |
| 相同点 | 形成磷酸二酯键 | 形成磷酸二酯键 |
修饰酶类
在基因克隆技术中,除了限制酶、连接酶、聚合酶这些主要的工具酶外,还经常使用某些酶的相关功能对DNA或RNA进行分子修饰,以便使基因工程操作更加巧妙、简便和高效。把这类酶统称为修饰酶。
其他基因工程常用酶:
- 核酸酶 SⅠ
- 碱性磷酸酶
- 核糖核酸酶 A
- 核糖核酸酶 H
- 蛋白酶 K
- 溶菌酶
核酸酶SⅠ
是一种单链特异的核酸酶,在最适的酶催反应条件下,降解单链 DNA 或 RNA,产生带磷酸的单核苷酸或寡核苷酸。对双链 DNA、双链 RNA 和 DNA-RNA 杂交体相对不敏感。通常水解单链 DNA 的速率要比水解双链 DNA 快 75000 倍。
S1 核酸酶的单链水解功能也可以作用对双链 DNA 中的一个切口发生作用,并从单链部位切断核酸分子。
S1 单链核酸酶的用途:使 S1 核酸酶在分析核酸杂交分子(RNA-DNA)的结构、给 RNA 分子定位、测定真核基因中间隔子序列的位置、去除 DNA 片段中突出的单链尾,以及打开在双链 cDNA 合成期间形成的发夹环起作用
碱性磷酸酶
其活性是催化核酸脱掉磷酸基团,从而使 DNA(或RNA)片段的 5′ 磷酸末端转换成 3′ 羟基末端,即所谓的核酸分子的脱磷酸作用。主要作用是防止 DNA 片段自连。
核糖核酸酶 A
核糖核酸酶 A(RNase A)对核糖核酸有水解作用,但对脱氧核糖核酸则不起作用。广泛用来去除 DNA 制品中的污染 RNA。
核糖核酸酶 H
核糖核酸酶 H(Rnase H):它能够特异性地水解杂交到 DNA 链上的 RNA 磷酸二酯键,故能分解 RNA/DNA 杂交体系中的 RNA 链。主要用于在 cDNA 克隆合成第二链之前去除 RNA。
蛋白酶 K
是一种强力蛋白溶解酶,是 DNA 提取的关键试剂。该酶 在较广的 pH 范围(4 ~ 12.5)内及高温(50 ~ 70°C)均有活性,在 DNA 提取中,主要作用是酶解与核酸结合的组蛋白,使 DNA 游离在溶液中,随后用不同方法进行抽提,除去杂质,收集 DNA。
溶菌酶
是一类水解细菌细胞壁中肽聚糖的酶,破碎细菌细胞壁,主要用于提取质粒和原核生物 DNA。
第三章 基因工程载体
载体(Vectors):在基因工程操作中,把能携带外源 DNA 或基因进入宿主细胞的 DNA 分子叫载体。↩
理想基因工程对载体的要求:
- 在宿主细胞内能独立复制。
- 有选择性标记,用于筛选。
- 具备合适的酶切位点,外源 DNA 插入其中不影响载体的复制。
- 分子量小,拷贝数多。
- 容易从宿主细胞中分离纯化。
载体的功能:
- 运送外源基因高效转入受体细胞
- 为外源基因提供复制能力或整合能力
- 为外源基因的扩增或表达提供必要的条件
质粒载体
质粒(plasmid):是独立于染色体以外的能自主复制的双链闭合环状 DNA 分子。存在于细菌、霉菌、蓝藻、酵母等细胞中。↩
质粒的一般特性
- 分子小:1 ~ 200 kb
- 编码基因少:2 ~ 3 个中等大小的蛋白质。如抗菌素抗性、代谢特征等
- 环形状:双链环状 DNA。(酵母的“杀伤质粒”是 RNA)
- 质粒的三种空间构型:
- 共价闭合环状 DNA(cccDNA),呈超螺旋(SC)(super coil)
- 开环 DNA(open circular,ocDNA),一条链上有一至数个缺口。
- 线形 DNA(linear,lDNA)
- 质粒空间构型与电泳速率
质粒 DNA 的生物学特性
- 不相容性(不亲和性):有相同复制起始区的不同质粒不能共存于同一宿主细胞中。该不相容性的分子基础主要是由于它们在复制功能之间的相互干扰造成的。
- 复制类型:
- 严谨型质粒的复制受到宿主细胞蛋白质合成的严格控制
- 松弛型质粒的复制不受宿主细胞蛋白质合成的严格控制。
- 质粒转移性,在大肠杆菌中的质粒,可以分为:
- 接合型质粒,能自我转移,如 F 质粒(F 因子)
- 非接合型质粒,不能自我转移,如:R 质粒(抗性质粒)
大肠杆菌接合(conjunction):F 因子携带基因编码的蛋白质能使两个细菌间形成纤毛状细管连接的接合(conjugation),通过这细管遗传物质可在两个细菌间传递。
组建理想质粒载体必须具备的条件,见 理想载体至少必备的条件有哪些
抗菌素选择原理:不带有抗菌素抗性基因的受体菌不能在含有抗菌素的培养基(选择培养基)中生长。当带有抗菌素抗性基因的载体进入受体菌后,受体菌才能生长。
TODO 蓝白斑试验、乳糖操纵子调节机制
质粒载体的改造
- 去掉不必要的 DNA 区段。
- 减少限制酶的识别位点,一种酶只保留一个。(单一的限制性酶切位点)。
- 加入易于捡出的选择性标记基因。
- 对质粒进行安全性改造,要求质粒不能随便转移。
- 改造或增加基因表达的调控序列。
常用的质粒载体
pBR322,松弛型复制,拷贝数 50 ~ 100 / cell,用于基因克隆
pUC18/19,拷贝数 500 ~ 700/cell,装有多克隆位点(MCS),正选择颜色标记 lacZ’,用于基因克隆和测序
重组子的检测:pUC18 质粒结构中具有来自大肠杆菌 lac 操纵子的 lacZ’ 基因,所编码的 α-肽链可参与 α-互补作用。因此,在应用 pUC18 质粒为载体的重组实验中,可用 X-gal 显色法一步实现对重组子克隆的鉴定。
噬菌体载体
噬菌体是一类细菌病毒(Bacteriophage)
噬菌体的结构:蛋白质外壳内包裹着 DNA(双链、单链、线性、环状等)。
λ DNA 分子的特点:
- 长度为 48502 bp;
- 双链线性 DNA;
- λ 噬菌体包装 DNA 的大小为原 λ DNA 长度的 78 ~ 105%(38 ~ 54 kb)
- 两端有 cos 位点,可以环化。
cos 位点(cohensive-end site):λ DNA 两端各有 12 bp 的粘性末端,粘性末端形成的双链区域称为 cos 位点。
噬菌体的生活周期:
- 溶菌周期(裂解循环),烈性噬菌体(virulent phage)
- 溶原周期,温和噬菌体(temperate phage)感染细菌后,将自己的 DNA 整合到细菌的染色体 DNA 中。形成这一过程称为溶源化
烈性噬菌体溶菌生长的基本过程:
- 吸附,吸附到位于感染细胞表面的特殊接受器上。
- 注入,噬菌体 DNA 穿过细胞壁注入寄主细胞。
- 转变,被感染的细胞成为制造噬菌体颗粒的场所。
- 合成,大量合成噬菌体特有的核酸和蛋白质。
- 组装,包装了 DNA 头部和尾部组装成噬菌体的颗粒。
- 释放,合成的子代噬菌体颗粒从寄主细胞内释放出来。
向溶原和裂解方向的转变由感染复数和细胞的营养状态决定的,感染复数越高和营养状态越差,向溶原方向的频率就越高。
λ 噬菌体的基因组结构
λ 噬菌体基因组至少可编码 30 个基因,根据功能不同分为 3 个区:
- Nul 基因到 J 基因:其产物用于噬菌体 DNA 的包装和噬菌体颗粒的形成。
- J 基因到 gam 基因:编码基因调节、溶原状态的发生和维持以及遗传重组所需基因,其中许多基因是非必需,在构建载体是可以去掉,用外源 DNA 替代。
- gam 基因至 Rz 基因:包含噬菌体复制和裂解宿主所需的基因。

噬菌体载体改造
λ-DNA 载体的构建(一):删除重复的酶切位点
- 野生型的 λ-DNA 链上有 5 个 EcoRⅠ 位点和 7 个 HindⅢ位点,不利于重组操作,必须删除至 1 ~ 2 个
- 为了便于各种来源的 DNA 片段的克隆,还需要增加一些单一的酶切位点
- 除了简单的切割外,还需要采用定点突变技术去除或增添酶位点
λ-DNA 载体的构建(二):加装选择标记,与质粒不同,野生型 λ-DNA 上缺少合适的选择标记,因此加装选择标记是 λ-DNA 克隆载体构建的重要内容
λ-DNA 克隆载体上的选择标记主要有下列两类:
- 免疫功能类标记
- 颜色反应类标记
λ 噬菌体载体的两种类型:
- 置换型(replacement),可被外源 DNA 置换 λ 噬菌体非必需区的载体。两侧有一对限制性酶切位点的载体。插入外源 DNA 片段一般为 6 kb,最大 11 kb
- 插入型(insertion),含一个限制性位点可供插入外源 DNA 的载体,这类 λ 噬菌体载体称插入型载体。非必需区已经去除。插入外源 DNA 片段大小为 9 ~ 23 kb
克隆原理及步骤,结合 λ 包装过程
- 通过裂解过程增殖载体、回收、纯化
- 酶切、载体与外源 DNA
- 连接
- 体外包装
- 感染/铺平板
- 筛选
λ-DNA 重组分子的体外包装:
λ-DNA 重组分子需在体外人工包装成有感染力的噬菌体重组颗粒,方可高效导入受体细胞。
用于体外包装的蛋白质可直接从感染了 λ 噬菌体的大肠杆菌中提取,现已商品化。这些包装蛋白通常分为相互互补的两部分:一部分缺少 E 组份,另一部分则缺少 D 组份。包装时,当且仅当这两部分包装蛋白与重组 λ-DNA 分子混合后,包装才能有效进行,任何一种蛋白包装液被重组 λ-DNA 污染后,均不能被包装成有感染力的噬菌体颗粒,这也是基于安全而设计的。
λ-DNA 载体的优点:比一般的质粒载体的容量大的多。用在真核生物基因组文库的建立。
第四章 人工染色体载体
常规载体在工作时都是在不影响质粒或噬菌体复制功能的基础上装载外源 DNA 片段的,同时保持质粒或噬菌体的基本特性。但这些载体体所装载的容量有限。利用染色体的复制元件来驱动外源DNA片段的载体称为人工染色体载体,其装载外源 DNA 片段的容量可以与染色体的大小媲美。
人工染色体载体:利用染色体的复制元件来驱动外源 DNA 片段复制的载体称为人工染色体载体。↩
模拟染色体的复制方式,因此都能装载大片段的 DNA 片段。
目前常用的人工染色体载体包括:
- 黏粒载体(cosmid)
- 酵母人工染色体载体(YAC)
- 细菌人工染色体载体(BAC)
黏粒载体
黏粒载体(Cosmid)又叫柯斯质粒载体,这是一种由人工构建的含有 λ 噬菌体 DNA 的 cos 位点和质粒复制子的特殊类型的杂和质粒载体,具有质粒和噬菌体的双重特征,被称为 cosmid,指带有粘性末端位点的质粒。↩
柯斯质粒在结构组成上具有 λ 噬菌体的特性,也具有质粒载体的特性和高容量的克隆能力,一般可插入 30 ~ 45 kb 的外源基因,是构建真核生物基因文库的主要载体。
黏粒载体构成
- λ 噬菌体 DNA:cos位点
- 质粒:复制子、抗药性基因、多克隆位点
黏粒载体特点
- 含有噬菌体 DNA 的粘性末端 cos 位点,可像 λ 噬菌体 DNA 一样被噬菌体包装蛋白包装,能并高效导入受体细胞
- 具质粒复制起始位点,可像质粒一样在细菌中复制
- 带有抗药性基因,如 Amp r、Tet r,易筛选
- 具一个或多个限制酶酶切位点,插入外源 DNA
- 分子量较小(5 ~ 7 kb),但能容纳较大的 DNA 片段
与 λ 噬菌体 DNA 的差异
- 缺少噬菌体复制起点,进入细菌后不能像噬菌体一样复制;
- 除 cos 位点外,λ 基因组的其余部分都被切割掉,具有高容量的克隆能力,最大可插入 30 ~ 45 kb 的片段;
- 柯斯质粒进入细胞后,质粒的复制子进行复制。
克隆原理及步骤
- 分离外源 DNA 片段 30 ~ 45 kb
- 外源 DNA 与黏粒载体相连
- 体外包装:在 λ 的 A 蛋白的末端酶功能作用下切割 cos 位点,且 cos 方向相同,并将包装到成熟的 λ 噬菌体颗粒中去
- 感染 E.coli:线状的重组 DNA 被注入细胞,通过 cos 位点的环化形成黏粒载体,像质粒一样复制
酵母人工染色体
YAC 是酵母人工染色体(Yeast artificial chromosome)的缩写,是目前能容纳最大外源 DNA 片段(100 万碱基以上)的载体
把酵母染色体与基因复制和表达有关的主要部件都组装在质粒上,令质粒行使酵母的转录和复制功能
YAC 只以单抄本方式繁殖,所以不适用于以生产为目的的基因工程。要用于在酵母中构建大片段 DNA 文库特别用来构建高等真核生物的基因组文库,人类基因组研究和巨大基因如 DMD 基因达 Mbp(106 bp)数量级基因的克隆,并不用作常规的基因克隆。
YAC 的基本序列元件
YAC 载体为满足自主复制、染色体在子代细胞间的分离及保持染色体稳定的需要,必须含有以下元件:
- 酵母染色体 DNA 自主复制顺序(ARS):染色体自主复制起点, 如 ARS1 酵母复制起始子
- 酵母染色体的着丝粒顺序(CEN):保证酵母细胞分裂时染色体的分配
- 酵母染色体的端粒重复顺序(TEL):(两端各一个)维持染色体结构的稳定性,确保染色体完整性,维持染色体成为线状,保护染色体末端免受核酸酶侵袭
- 选择标记:用于重组克隆的筛选。在细菌和酵母菌中选择的标记基因,含有 Amp 大肠杆菌筛选标记;TRP1 色氨酸生物合成基因, URA3 尿嘧啶生物合成基因
- 克隆位点
YAC 载体的选择标记主要采用营养缺陷型基因:如色氨酸合成酶基因 trp 1和尿嘧啶合成酶基因 ura3 等
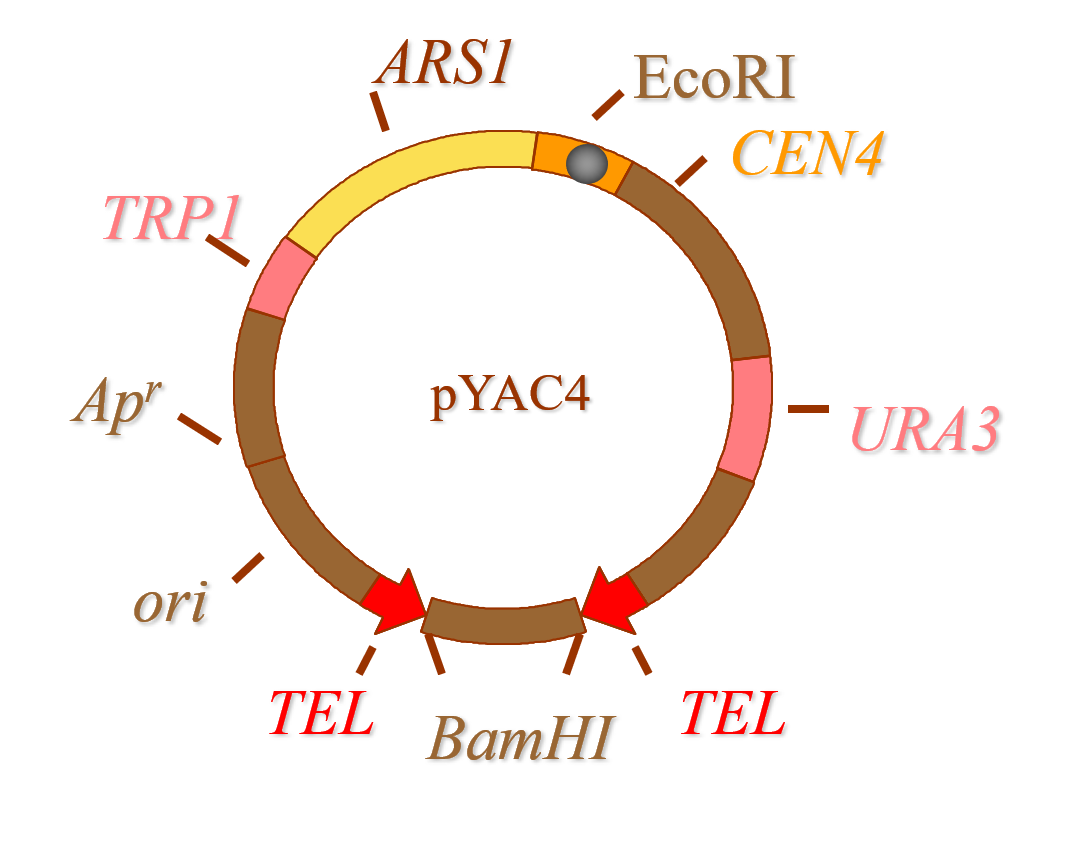
YAC 载体应含有下列元件:
- 酵母染色体的端粒序列(TEL)
- 酵母染色体的复制子(APS1)
- 酵母染色体的着丝粒序列(CEN4)
- 酵母系统的选择标记(URA3)
- 大肠杆菌的复制子标记(ori)
YAC 载体的装载量为:250 ~ 400 kb
YAC 载体的工作原理、主要用途以及其缺点,见 简述 YAC 载体的工作原理、应用以及其缺点
细菌人工染色体载体
细菌人造染色体通常是在大肠杆菌性因子 F 质粒的基础上构建的,其装载量范围在 120 ~ 300 kb 之间。
- 基于细菌性因子(F 因子)的复制点和氯霉素抗性标记基因,在 F 质粒(pMBO131)基础上构建了高容量的第一代 BAC 人工染色体(300 kb)
- BAC 载体在重组缺陷型(REC-)宿主菌只有 1 个拷贝,有利于保持 DNA 大分子,尤其是重复序列多的 DNA 大分子,在细胞内稳定复制而不发生重组
- F 因子(100 kb)又称致育因子,在细菌细胞内能自我复制,能整合到宿主染色体中。因其低拷贝特性使其重排和嵌合程度低。且 F 因子呈闭环结构,可以用常规技术从大肠杆菌中分离
细菌人工染色体的结构
- F 因子的 parA,parB,parC 基因以保证单拷贝的 BAC 质粒在大肠杆菌分裂时均匀分配到子代细胞
- 起始 oriS 基因,严密控制,决定低拷贝和起始
- 解旋酶基因 repC,易于 DNA 复制和决定复制方向
- 选择标志基因 Cmr 氯霉素抗性基因
- lacZ 基因,颜色鉴别重组子,外源基因插入其中
- loxP 和 cosN 位点,易于克隆 DNA 回收和操作
- 在多克隆位点两侧,还有 T7 及 SP6 启动子位点,用于制备 RNA 分子,进一步分析克隆的基因表达,作为染色体步行的探针,以及序列测定克隆的片段
BAC 载体与质粒载体的异同点
BAC 载体的工作原理与常规的质粒载体相似。
不同的是:
- BAC 载体装载的是大片段 DNA,一般在 100 Kb ~ 300 Kb 。
- BAC 载体的拷贝数小,制备难度大。
BAC 基因组文库的构建
- BAC 载体的制备:BAC DNA的分离、BAC DNA经酶切(Hind Ⅲ)线性化、去磷酸化反应
- 高分子量基因组 DNA 的制备:大分子量 HindⅢ 酶切片段的制备、脉冲电泳选择酶切片段
- BAC 克隆
- 电转化
- BAC 克隆鉴定
BAC 载体用途
各种类型的 pBACs 在大肠杆菌受体菌只能维持单一拷贝 pBACs 主要适用于:
- 克隆大型基因簇(gene cluster)结构
- 构建动植物基因文库
载体的种类和特征

四种常用载体的比较

第五章 核酸提取及电泳技术
DNA 提取方法
DNA 提取的几种方法:
- 质粒 DNA 的提取:碱裂解法、煮沸法
- 基因组 DNA 的提取:CTAB 法、SDS 法、苯酚抽提法
质粒 DNA 的提取
碱裂解法和煮沸法抽提质粒 DNA 原理,见 碱裂解法和煮沸法抽提质粒 DNA 的原理

基因组 DNA 的提取 —— CTAB 法
CTAB 法原理(植物 DNA 提取经典方法)
CTAB(hexadecyltrimethylammonium bromide,十六烷基三甲基溴化铵),是一种阳离子去污剂,可溶解细胞膜,并与核酸形成复合物。
该复合物在高盐溶液中(> 0.7 mol/L NaCl)是可溶的,通过有机溶剂抽提,去除蛋白、多糖、酚类等杂质后加入乙醇沉淀即可使核酸分离出来。
注:CTAB 溶液在低于 15℃ 时会形成沉淀析出,因此在将其加入冰冷的植物材料之前必须预热至 60℃ ,且离心时温度不要低于 15℃。
CTAB 提取缓冲液的经典配方:
| 组份 | Tris-HCl(pH 8.0) | EDTA (pH8.0) | NaCl | CTAB | β-巯基乙醇 |
|---|---|---|---|---|---|
| 终浓度 | 100 mM | 20 mM | 1.4M | 2%(W/V) | 0.1%(V/V)使用前加入 |
- Tris-HCl(pH 8.0)提供一个缓冲环境,防止核酸被破坏;
- EDTA 螯合 Mg2+ 或 Mn2+ 离子,抑制 DNase 活性;
- NaCl 提供一个高盐环境,使 DNA 充分溶解,存在于液相中;
- CTAB 溶解细胞膜,并结合核酸,使核酸便于分离;
- β-巯基乙醇是抗氧化剂,有效地防止酚氧化成醌,避免褐变,使酚容易去除

基因组 DNA 的提取 —— SDS 法
SDS 法原理:
- SDS 是一种离子型表面活性剂,在高温(55 ~ 65℃)条件下能溶解细胞膜和核膜蛋白,裂解细胞膜和核膜,使染色体离析,蛋白变性,释放出核酸;
- 上清液中的 DNA 用酚/氯仿抽提,反复抽提后用乙醇沉淀水相中的 DNA。

DNA 提取的基本步骤
- 材料准备
- 破碎细胞或包膜-内容物释放
- 核酸分离、纯化
- 沉淀或吸附核酸,并去除杂质
- 核酸溶解在适量缓冲液或水中
材料准备:
基因组 DNA 的提取
- 最好使用新鲜材料,低温保存的样品材料不要反复冻融
- 提取血液基因组 DNA 时,要选择有核细胞(白细胞)
- 组培细胞培养时间不能过长,否则会造成 DNA 降解
- 含病毒的液体材料 DNA 含量较少,提取前先富集
质粒 DNA 的提取
- 使用处于对数期的新鲜菌体(老化菌体导致开环质粒增加)
- 培养时应加入筛选压力,否则菌体易污染,质粒易丢失
- 尽量选择高拷贝的质粒,如为低拷贝或大质粒,则应加大菌体用量
- 菌株不要频繁转接(质粒丢失)
细胞裂解:
基因组 DNA 的提取
- 材料应适量,过多会影响裂解,导致DNA量少,纯度低
- 针对不同材料,选择适当的裂解预处理方式:植物材料(液氮研磨)、动物组织(匀浆或液氮研磨)、组培细胞(蛋白酶 K)、细菌(溶菌酶破壁)、酵母(破壁酶或玻璃珠)
- 高温温浴时,定时轻柔振荡
质粒 DNA 的提取
- 菌体量适当
- 培养基去除干净,同时保证菌体在悬浮液中充分悬浮
- 变性的时间不要过长(5分钟),否则质粒易被打断
- 复性时间也不宜过长,否则会有基因组 DNA 的污染
- G+菌、酵母质粒的提取,应先用酶法或机械法处理破壁
基因组 DNA 的提取和质粒 DNA 的提取的核酸分离、纯化:
- 采用有机(酚/氯仿)抽提时应充分混匀,但动作要轻柔
- 酚/氯仿抽提去除蛋白质
- 在待沉淀溶液中加入 1/2 体积的 5 M NaCl,高盐可溶解多糖,去除多糖
- 70% 的乙醇洗涤去除盐离子
DNA 的定量和纯度测定
- 紫外分光光度计测定
- 琼脂糖凝胶电泳估算
紫外分光光度计测定
- DNA/RNA 在 260 nm 处有最大的吸收峰
- 蛋白质在 280 nm 处有最大的吸收峰
- 盐和小分子则集中在 230 nm 处
测浓度:对标准样品来说,浓度为 1 μg/ml 时,DNA 钠盐的 OD260 = 0.02,当 OD260 = 1 时,dsDNA 浓度约为 50 μg/ml。DNA(mg/ml)= 50 × OD260 读数 × 稀释倍数 / 1000
测纯度:DNA/RNA 纯品的 OD260 / OD280 为 1.8 左右,故根据 OD260 / OD280 的值可以估计 DNA 的纯度。
- 若比值大于 1.9 说明含有 RNA
- 比值小于 1.6 说明有残余蛋白质、酚类等存在。
琼脂糖凝胶电泳估算:EB 能嵌入 DNA 平面结构,在 300 nm 的紫外光照下能发色红光。荧光强度与 DNA 含量及大小成正比。通过与已知 Marker 条带的亮度对比来估计目的 DNA 的含量。
电泳技术
电泳的概念:带电物质在电场中向相反电极移动的现象称为电泳(electrophoresis)。
电泳现象早在十九世纪初就已发现(1808 年俄国物理学家 Reŭss 进行了世界上第一次电泳实验)。但电泳技术的广泛应用,则是在 1937 年用滤纸作为支持介质成功地进行纸电泳以后,特别是近几十年以来,电泳技术发展很快,各种类型的电泳技术相继诞生,在生物化学、医学、免疫学等领域得到了广泛应用。
琼脂糖凝胶电泳:琼脂糖凝胶孔径相对较大,其分辨率较低,一般用于核酸的分离分析。
聚丙烯酰胺凝胶电泳:其分辨率较高,可用于核酸和蛋白质的分离、纯化及检测。
电泳的基本原理
在一定 pH 条件下,每一种分子都具有特定的电荷(种类和数量)、大小和形状,在一定时间内它们在相同电场中泳动速度不同,各自集中到特定的位置上而形成紧密的泳动带。这就是带电粒子可以用电泳技术进行分离、分析和鉴定的基本原理。
琼脂糖凝胶电泳
琼脂糖凝胶电泳是用琼脂或琼脂糖作支持介质的一种电泳方法。
琼脂糖凝胶约可区分相差 100 bp 的 DNA 片段,其分辨率虽比聚丙烯酰胺凝胶低,但它制备容易,分离范围广,尤其适于分离大片段 DNA。普通琼脂糖凝胶分离 DNA 的范围为 0.2 ~ 20 kb。
安全的核酸染料:溴化乙啶(EB)是一种最常用的物美价廉的核酸染料,有致癌性。因此,近年来数种较为安全的核酸染料在逐步取代 EB 的应用,如 SYBRGreen、GeneFinder 和 GoldView 核酸荧光染料,GeneFinder 和 Glod View 染色后的条带可以在紫外灯下观察、照相,在可见光(400 ~ 500 nm 的蓝绿光)透射仪下观察条带呈绿色。
琼脂糖凝胶中 RNA 的检测:RNA 分析前需要变性,加入甲醛变性剂,使 RNA 分子内的氢键打开,除去二级结构。
影响 DNA 迁移率的因素:
- DNA 分子的大小、构象,凝胶浓度,电压,电泳缓冲液的组成等
- DNA 带负电荷,迁移率与分子量对数成反比,DNA 片段越长,泳动速度越慢
- DNA 分子构象影响迁移率:共价闭环 DNA > 线状 DNA > 开环 DNA
- 泳动速度与凝胶浓度成反比
- 在低电压时,泳动速度与电场强度成正比
- 电泳缓冲液的组成及其离子强度影响 DNA 的电泳迁移率。
聚丙烯酰胺凝胶电泳
聚丙烯酰胺凝胶电泳(polyacrylamide gel electropho-resis,PAGE)是以聚丙烯酰胺凝胶作为支持介质的一种电泳方法。是由丙烯酰胺单体(简称Acr)和交联剂 N,N-甲叉双丙烯酰胺(简称 Bis)在催化剂作用下合成的。Acr 和 Bis 两种都有神经毒性,需带 PE 手套。
凝胶孔径大小,主要受凝胶浓度的影响。凝胶浓度越大,孔径越小。凝胶浓度过大,胶硬而脆,易折断;浓度过小,凝胶稀软,不易操作,也易断裂。
聚丙烯酰胺凝胶电泳可很好分辨 100 ~ 1000 bp,其分辨率可达 1 bp。
聚丙烯酰胺凝胶电泳:
- 非变性:检测 DNA(PAGE)
- 变性:检测蛋白质(SDS-PAGE)
PCR 技术及其应用
体外扩增特异 DNA 片段的技术,能快速、特异地扩增目的 DNA 片段。能通过试管内的数小时反应将特定的 DNA 片段扩增数百万倍。迅速获取大量的单一核酸片段,为分子生物学研究提供了强大的工具。
PCR 的原理
- 由一对引物介导,通过温度的调节,使双链 DNA 变性为单链 DNA、单链 DNA 与引物复性(退火)成为引物-DNA 单链复合物、以及在 dNTPs 存在下 DNA 聚合酶使引物延伸而成为双链 DNA(引物的延伸);
- 这种热变性-复性-延伸的过程,就是一个 PCR 循环;
- 一般通过 20 ~ 30 个循环之后,就可获得大量的要扩增的 DNA 片段。
PCR 反应体系
PCR 反应体系包含:
- 模板 DNA
- Taq 酶(Taq DNA polymerase)
- dNTP
- 引物(Primers)(上游、下游)
- 缓冲液体系(buffer)
- Mg2+
- ddH2O
- 模板变性 (Denaturation),双链 DNA 模板在 95℃ 变性为单链 DNA。
- 引物退火 (Annealing),引物与单链 DNA 互补并退火。
- 延伸反应 (Extention) ,耐热的 DNA 聚合酶催化子链的合成。
PCR 反应条件的设定
待扩增片段:1000 bp
引物 Tm = 37 ~ 72℃
DNA 模板变性: 94℃, 5分钟;
PCR 循环 (30 次): 94℃, 30秒; 50℃, 30秒; 72℃, 1分钟;
最终延伸: 72℃, 7 ~ 10分钟
引物(寡核苷酸引物)
引物:引物是 PCR 特异性反应的关键,PCR 产物的特异性取决于引物与模板 DNA 互补的程度。理论上,只要知道任何一段模板 DNA 序列,就能按其设计互补的寡核苷酸链做引物,利用 PCR 就可将模板 DNA 在体外大量扩增。
引物是与待扩增DNA片断两翼互补的一段特异的寡核苷酸片断。
引物的作用:决定 PCR 扩增产物的特异性与长度,决定 PCR 反应的成败。
引物设计的原则,见 引物设计需遵循的原则有哪些
PCR 的类型
- 标准 PCR
- 长 PCR (Long PCR):PCR 产物 > 5 kb
- 巢式 PCR (Nested PCR)
- 反向 PCR (Inverse PCR)
- 菌落 PCR (Colony PCR)
- 逆 (反) 转录 PCR (RT-PCR)
- 实时定量 PCR (Real Time Quantitative PCR, qRT-PCR)
反向 PCR (reverse PCR) :用反向的互补引物来扩增两引物以外的 DNA 片段,对某个已知 DNA 片段两侧的未知序列进行扩增。可对未知序列扩增后进行分析,如探索邻接已知 DNA 片段的序列;用于仅知部分序列的全长 cDNA 的克隆, 建立基因组步移文库。↩
实时荧光 PCR 技术:通过荧光染料或荧光标记的特异性的探针,对 PCR 产物进行标记跟踪,实时在线监控反应过程;结合相应的软件可以对结果进行分析,计算待测样品的初始模板量。
PCR 的应用领域
- 生物学基础研究:目的基因扩增和鉴定;DNA序列测定;定点突变;基因表达分析
- 医学临床应用:遗传疾病基因诊断;致病病原体检测;癌基因检测;器官移植组织配型
- 法医学物证鉴定:个体识别;亲子鉴定
- 其他:动、植物检疫(转基因动植物检测);生物物种鉴定,系统进化研究;分子考古学(恐龙 DNA 分析)等
第六章 基因工程的常规技术之分子杂交
分子杂交:是一类核酸和蛋白质分析方法,用于检测混合样品中特定核酸分子或蛋白质分子是否存在,以及其分子量的大小。↩
根据其检测对象不同,可分为:
| 分子杂交 | 检测对象 | 探针 |
|---|---|---|
| Southern 杂交 | DNA | 核酸 |
| Northern 杂交 | RNA | 核酸 |
| Western 杂交 | 蛋白质 | 抗体 |
以及由此而简化的斑点杂交、菌落杂交和原位杂交等。
核酸分子杂交的基本原理
带有互补核苷酸序列的单链 DNA 或 RNA,当它们混合在一起时,其相应的同源区段将会退火形成双链的结构,形成杂种核酸分子。
当用一个标记的核酸分子与核酸样品杂交,便可查明该样品中是否存在与该标记核酸分子具有同源性的核酸分子。
这个标记的核酸分子称为探针(probe),可以是 DNA,RNA 或合成的寡核苷酸。
用合适方法(如放射自显影或免疫组织化学等技术)把标记物检测出来,就可确定靶核苷酸序列的拷贝数及表达丰度等。
分子杂交基本过程
- 核酸印迹(Nucleic acid blotting):将核酸样品(DNA、RNA或蛋白质)在凝胶上分离,然后将样品通过影印的方式转移到固相支持物如滤膜上。
- 核酸杂交(Hybridization):将印迹有核酸样品的滤膜同带有放射性标记或其它标记的 DNA 或 RNA 探针(Probe)或抗体进行杂交。
- 结果检测(Detection Results):通过放射自显影或显色反应,判断样品中是否有与探针同源的核酸分子或与抗体反应的蛋白质分子。
Southern 杂交
Southern 杂交原理:通过毛细管作用、电转移、真空转膜等方法,使在凝胶电泳中已分离的 DNA 片段转移并结合到适当的滤膜上,然后通过同已标记的探针进行杂交,以检测被转移 DNA 片段,称为 DNA 印迹杂交技术。
在 1975 年,由英国的 E.Southern 首先设计发明的,因此又称为 Southern 杂交(Southern blotting)。
Southern 杂交特点:
- 作用对象:DNA 分子
- DNA 片段需经凝胶电泳分离
- 固相支持物:可选用的滤膜种类较多
- 灵敏度高
Southern 杂交过程:
- DNA 的片段化(限制性酶切)及其电泳分离
- 凝胶电泳后的变性处理,在 0.4 mol/L NaOH 碱性条件下变性,使双链 DNA 分子变成单链。再在 1.5 mol/L NaCl、1 mol/L Tris(pH 7.4)条件下中和使 DNA 仍保持单链状态。
- 转移并固定到滤膜上,通过毛细管渗吸或电转移或真空转移的方式,将凝胶上的 DNA 转移到硝酸纤维素滤膜或尼龙膜上。最后通过 80℃ 处理或紫外线照射将 DNA 固定在滤膜上。 转膜方法:毛细管虹吸印迹法、电转印法、真空转移法
- 探针的制备及杂交
- 预杂交:将结合了 DNA 分子的滤膜先与特定的预杂液进行预杂交,也就是将滤膜的空白处用鱼精 DNA 或小牛血清蛋白封闭起来,防止在杂交过程中滤膜本身对探针的吸附。
- 标记与目的序列同源的核苷酸片段作为探针
- 杂交:在一定的溶液条件和温度下,将标记的核酸探针与滤膜混合,如果滤膜上的 DNA 分子存在与探针同源的序列,那么探针将与该分子形成杂合双链,从而吸在滤膜上。
- 洗膜:经过一定的洗涤程序将游离的探针分子除去。
- 检测与分析
- 通过放射自显影或生化检测,就可判断滤膜上是否存在与探针同源的 DNA 分子及其分子量。
- Southern 杂交主要用来判断某一生物样品中是否存在某一基因(DNA 水平)。
- Southern 杂交也可检测目的基因的拷贝数。

常用的固相支持物:
- 硝酸纤维素膜:优点是吸附能力强,杂交信号本底低。缺点是 DNA 分子结合不牢固,脆性大易破裂
- 尼龙膜:优点是结合单链,双链 DNA 的能力比硝酸纤维素膜强;缺点:杂交信号本底高
- 化学活化膜:优点:DNA与膜共价结合;对不同大小的DNA片段有同等结合能力;缺点:结合能力较上述两种膜低
Northern 杂交
Northern 杂交原理:根据毛细管作用的原理,使在凝胶电泳中已分离的 RNA 转移并结合到适当的滤膜上,然后通过同已标记的探针进行杂交,以检测被转移 RNA 片段,称为 RNA 印迹杂交技术。
在 1979 年,由 J. C. Alwine 等人设计,也称为 Northern 杂交(Northern blotting)。
Northern 杂交特点:
- 作用对象:RNA 分子
- 需经凝胶电泳分离
- 固相支持物:尼龙膜为主,结合牢固程度高
- 主要用于基因是否表达以及基因表达强弱的检测(RNA 水平)
Northern 印迹与 Southern 印迹的不同
- 转移的对象不同,Northern 印迹是将 RNA 变性及电泳分离后,将其转移到固相支持物上的过程。
- 虽然 RNA 电泳前不需像 DNA 那样进行酶切,但也需要变性。不过变性方法是不同的,它不能用碱变性,因为碱变性会导致 RNA 的降解
Western 杂交
Western 杂交原理:在电场的作用下将电泳分离的蛋白质从凝胶转移至一种固相支持体,然后用这种蛋白质的特异抗体来检测。
Western 杂交的总体过程也与 Southern 杂交相似,只不过在印迹转移过程中转移的是蛋白质而不是 DNA,这种将蛋白质样品从 SDS-PAGE 凝胶通过电转移方式转移到滤膜的方法,称为 Western blotting
其后的杂交过程不是真实意义的分子杂交,而是通过抗体(探针)以免疫反应形式检测滤膜上是否存在被抗体识别的蛋白质,并判断其分子量。所用的探针不是 DNA 或 RNA,而是针对某一蛋白质制备的特异性抗体。
Western 杂交特点:
- Western 杂交主要用来检测细胞或组织样品中是否存在能被某抗体识别的蛋白质,从而判断在翻译水平上某基因是否表达。(翻译水平)
- 这种检测方法与其它免疫学方法的不同是,一方面可以避免非特异性的免疫反应,而且更关键的是可以检测出目标蛋白质的分子量,从而直观的在滤膜上显示出目标蛋白。
Western Blot 流程
- 蛋白样品的制备
- SDS-PAGE 电泳
- 转膜
- 封闭
- 一抗杂交
- 二抗杂交
- 底物显色
斑点杂交
斑点杂交原理:在 Southern 杂交的基础上发展起来的用于快速检测特异核酸分子的杂交技术。将核酸样品直接点到适当的滤膜上,然后进行杂交检测。
斑点杂交优点:
- 快速简便,不需电泳分离
- 一次的检测量大
- 更适用于定量检测
菌落或噬菌斑杂交
菌落或噬菌斑杂交原理:
- 将菌落或噬菌斑直接转移到滤膜上,使溶菌变性的 DNA 同滤膜原位结合,然后进行杂交检测。
- 杂后根据杂交信号及其相对应的菌落或噬菌斑的位置,杂交便可从大量菌落或噬菌斑中筛选出含有目标序列(阳性克隆子)(与探针同源)的菌落或噬菌斑。
核酸杂交分类表
| 杂交方法 | 适用范围 |
|---|---|
| Southern 印迹 | 检测经凝胶电泳分开的 DNA 分子,需转印到膜上 |
| Northern 印迹 | 检测经凝胶电泳分开的 RNA 分子,需转印到膜上 |
| 斑点印迹杂交 | 检测未经分离的、固定在膜上的 DNA 或 RNA 分子 |
| 菌落或噬菌斑杂交 | 检测固定在膜上的、经裂解后从细菌或噬菌体中释放出的 DNA 分子 |
探针
在一个核酸样品中查找是否存在某一特定序列的分子可用分子杂交来检测,但首先要有一段与目标核酸分子的序列同源的核酸片段。
将该片段标记后与样品核酸进行分子杂交,通过检测标记核酸的存在从而判断样品中特定核酸片段的存在。
用作检测的核酸片段即为探针(probe)。
核酸探针的来源
- 基因组 DNA 探针
- cDNA 探针
- RNA 探针
- 寡核苷酸探针:人工合成、15 ~ 30 nt
探针的标记
- 均匀标记:间接标记不是标记探针分子本身,而是复制一段新的探针分子,在复制过程中掺入标记的核苷酸(如 [α-32P] dATP),从而使整个新分子被均匀地标记。
- 末端标记:直接将探针分子的某个原子替换为放射性同位素原子,或直接在探针分子上加入标记的原子或复合物,这种直接标记一般是在探针分子的末端进行标记,亦称末端标记。
常用的探针标记物
| 标记物种类 | 主要标记物 | 特点 |
|---|---|---|
| 放射性同位素标记物 | 32P、35S、3H | 灵敏度极高;不影响各种酶促反应;不影响碱基配对的特异性和稳定性;特异性高,假阳性少;污染环境;半衰期短; |
| 非放射性标记物 | 生物素、地高辛、荧光素 | 安全;稳定性好;灵敏度和特异性较低; |
核酸分子杂交信号的检测
- 放射性同位素标记探针:放射自显影
- 非放射性同位素标记探针:酶联免疫反应 + 显色反应
地高辛原理:地高辛 + 抗地高辛抗体(带有荧光素或酶的标记)
常用的探针标记法
- 切口移位(平移)法
- PCR 标记法
- 末端标记法
第六章 基因工程的常规技术之基因芯片
生物芯片:是将大量生物识别分子按预先设置的排列固定于一种载体(如硅片、玻片及高聚物载体等)表面,根据分子间的特异性相互作用的原理,如核酸杂交反应,抗原抗体反应等来检测各种生物分子存在的量的一种技术,以实现对细胞、蛋白质、核酸以及其它生物组分的准确、快速、大信息量的检测。
生物芯片包括:DNA 芯片、蛋白质芯片
DNA 芯片:是最重要的一种生物芯片,又称基因芯片,是一种高通量的斑点杂交技术,通过将大量的 DNA 分子固定于支持物上,并与标记的样品杂交,然后通过自动化仪器检测杂交信号来判断样品中靶分子的信息,诸如基因组 DNA 突变谱和 mRNA 表达谱的检测等。
基因芯片主要技术流程
- 芯片制备
- 样品荧光标记
- 芯片杂交
- 芯片洗涤和扫描
- 扫描图像分析
芯片的制备
(一)支持物的预处理
- 实性材料:硅芯片、玻片和瓷片。需进行预处理,使其表面衍生出羟基、氨基活性基团。
- 膜性材料:聚丙烯膜、尼龙膜、硝酸纤维膜。通常包被氨基硅烷或多聚赖氨酸
(二)探针的制备
- 原位合成法
- 接触点样法
- 喷墨法
探针的固化:打印探针后,需要将其固定在支持物表面,同时也要封闭支持物上未打印区域以防止核酸样品的非特异性固定
样品的准备
- 样品的分离纯化:DNA,mRNA
- 扩增:PCR,RT-PCR,固相 PCR
- 标记等过程:荧光标记(常用 Cy3、Cy5),生物素、放射性标记
分子杂交
样品与 DNA 芯片上的探针阵列进行杂交。与经典分子杂交的区别:
- 杂交时间短,30 分钟内完成
- 可同时平行检测许多基因序列
检测分析
- 激光激发使含荧光标记的 DNA 片段发射荧光
- 激光扫描仪或激光共聚焦显微镜采集各杂交点的信号
- 软件进行进行图象分析和数据处理
DNA 芯片的应用
- 科研方面的应用
- 表达谱分析
- SNP 分析(包括最新的 CSV/CNV分析,GWAS)
- CGH 分析
- Chip-ChIP 分析
- 临床相关应用
- 传染性疾病的检测(有无和耐药性检测)
- 遗传性疾病的检测
表达谱芯片
- 肿瘤相关基因(正常与肿瘤组织表达差异)
- 药物筛选(培养细胞药物刺激前后表达差异)
- 发育(同一组织不同发育时期基因表达差异)
- 组织发生(不同组织或器官的基因表达差异)
DNA 芯片未来展望
- 作为一门技术本身已经非常成熟。
- 今日的 DNA 芯片在表达谱分析,SNP 分析以及 Chip-ChIP 分析方面的地位将会逐渐被下一代测序技术所取代,但以其灵活性和高效性,在临床应用方面将长久的发挥作用。
- DNA 芯片发展过程中所积累起来设备,技术以及数据分析软件将为其它类型的高密度芯片技术的发展提供重要参考。
- 开发其它可能的应用:记住核心 addressable-decoder.
- ……
第六章 基因工程的常规技术之 DNA 序列分析
1977 年,Maxam 和 Gilbert 报道了化学降解法测定 DNA 的序列。
1977 年,Sanger 在引入双脱氧核苷三磷酸(ddNTP)后,形成了双脱氧链终止法,使得 DNA 序列测定的效率和准确性大大提高。
1980 年,Frederik Sanger 和 Walter Gilbert 因“在测定核酸碱基序列上的突出贡献”而分享了当年的诺贝尔化学奖。
第一代 DNA 测序技术
第一代 DNA 测序技术:传统的化学降解法、双脱氧链终止法以及在它们的基础上发展来的各种 DNA 测序技术。
第一代 DNA 测序技术包括:化学降解法、双脱氧链终止法、荧光自动测序技术和杂交测序技术。
化学降解法
将一个 DNA 片段的 5’ 端磷酸基作放射性标记,再分别采用不同的化学方法修饰和裂解特定碱基,打断磷酸二脂键,从而产生一系列长度不一而5’ 端被标记的 DNA 片段,这些以特定碱基结尾的片段群通过凝胶电泳分离,再经放射线自显影,确定各片段末端碱基,从而得出目的 DNA 的碱基序列。
碱基特异性化学切割反应:
- 硫酸二甲酯(DMS):使 DNA 分子中鸟嘌呤(G)上的 N7 原子甲基化。
- 哌啶甲酸:在酸性条件使 DNA 链脱嘌呤(G & A)。
- 肼:在碱性条件使 DNA 分子中胸腺嘧啶和胞嘧啶(T & C)的嘧啶环断裂;但高盐条件下,只 C 断裂,而不与 T 反应。
在不同的酸、碱、高盐和低盐条件下,三种化学试剂按不同组合可以特异地切割核苷酸序列中特定的碱基。
将标记 DNA 分为 G、GorA、CorT 和 C 4 个反应体系
测定 DNA 长度大约为 250 bp。
| 反应体系 | 碱基修饰试剂 | 碱基修饰反应 | 断裂点 |
|---|---|---|---|
| G | 硫酸二甲酯 | 鸟嘌呤甲基化 | G |
| G + A | 哌啶甲酸 | 脱嘌呤作用 | G 和 A |
| C+T | 肼 | 嘧啶开环 | C 和 T |
| C | 肼(加盐) | 胞嘧啶开环 | C |
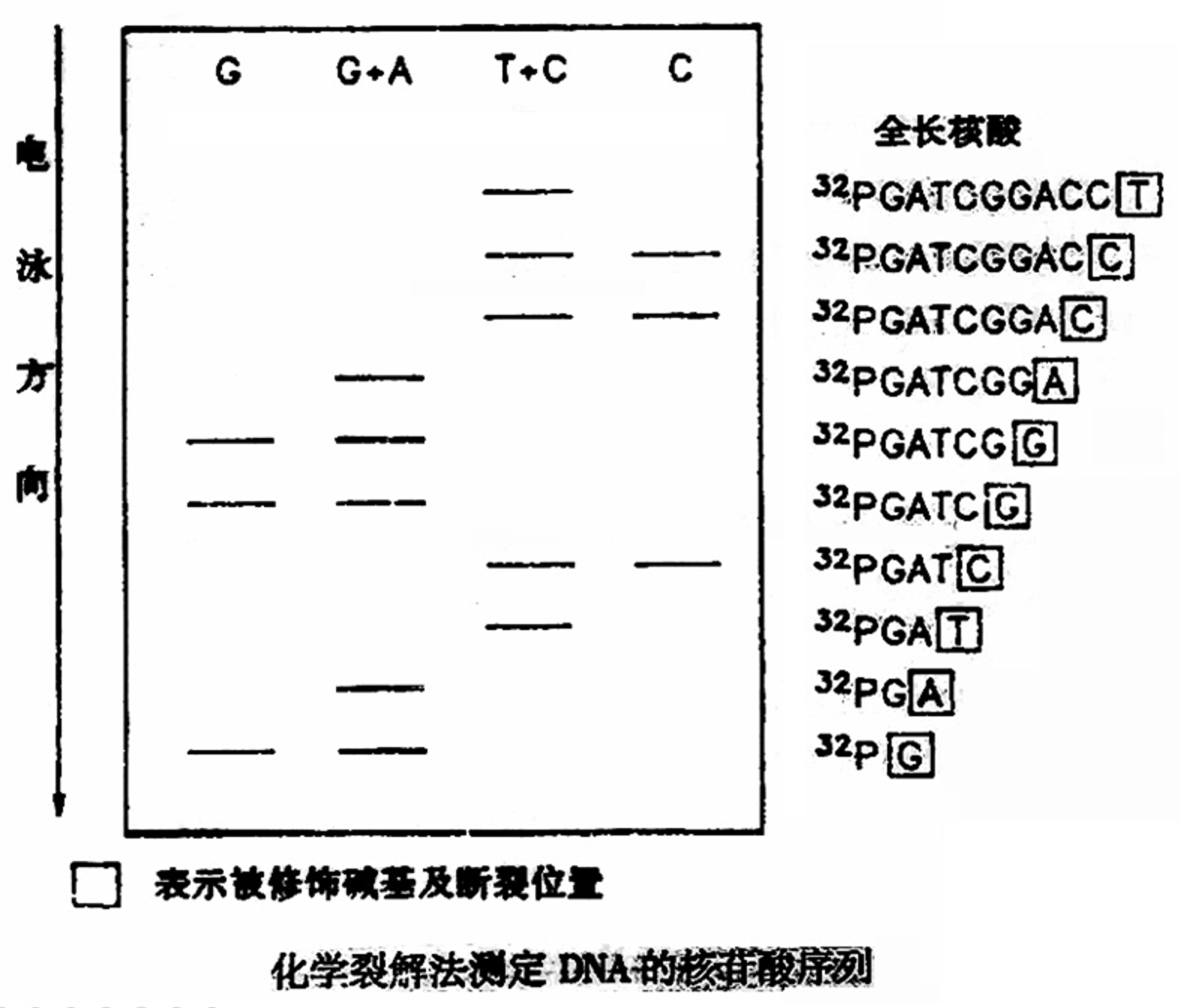
化学降解法刚问世时,准确性较好,也容易为普通研究人员所掌握,因此用得较多。但化学降解法操作过程较麻烦,且用到放射性物质,逐渐被简便快速的 Sanger 法所代替。
双脱氧链终止法(Sanger法)
1977年,英国人 Fred Sanger 发现,如果在 DNA 复制过程中掺入 ddNTP,就会产生一系列末端终止的 DNA 链,并能通过电泳按长度分辨。不同末端终止 DNA 链的长度是由掺入到新合成链上随机位置的 ddNTP 决定的。
Sanger 双脱氧链终止法:
- 与 PCR 反应类似。
- 反应体系中包含:模板 DNA, Taq 酶, dNTPs, ddNTPs 和测序引物;
- 反应过程:变性-复性-延伸-终止
Sanger 法是在反应体系中加入 2’,3’-ddNTP,由于其没有 3’-OH 而不能与下一个核苷酸相连,于是 DNA 链的合成便终止。
Sanger 法:
- 在 PCR 时加入标记的复制终止剂,比如 ddA,ddT,ddC,ddG(相应于 4 种碱基)
- ddNTP 的两个作用:可以当作正常碱基参与复制、一旦链入 DNA 中,其后就不能再继续连接
- 电泳
- 谁终止,碱基就是谁
Sanger 双脱氧链终止法
- 一个样品需要 4 个合成反应(每个反应分别加 4 种 dNTP 和一种 ddNTP)
- 反应结束后,分 4 个泳道进行聚丙烯酰胺凝胶电泳分离
- 各自泳道按照分离片段大小进行读数
DNA 测序自动化和大规模测序
特点:
- 原理同 sanger 法
- 标记物为荧光染料(与双脱氧核苷三磷酸共价相连)
- 激光扫描自动测序
- 结果清晰、准确、分辨率高
- 测序速度快
毛细管电泳基本原理:与链终止法测序原理相同,只是用不同的荧光色彩标记 ddNTP,如 ddATP 标记红色荧光, ddTTP 标记绿色荧光,ddCTP 标记蓝色荧光, ddGTP 标记黄色荧光,由于每种 ddNTP 带有各自特定的荧光颜色,而简化为由 1 个泳道同时判读4种碱基。
DNA 自动测序步骤:
- 4 种带有不同荧光染料标记的终止物 ddNTPs
- Sanger 测序反应
- 反应产物毛细管电泳分离
- 激光激发、荧光信号采集、计算机分析与 DNA 自动排序
DNA 自动测序与手工测序的不同点
- 标记物不同:手工测序采用放射性核素标记,而自动测序采用 4 种荧光染料分别标记 ddNTP 或标记引物
- 加样方式不同:手工测序,一个样品的 4 个测序反应物分别在不同泳道进行,而自动测序可在一个泳道内电泳
- 检测手段不同:手工测序采用放射自显影,从 4 种寡聚核苷酸的梯子形图谱中读出 DNA 序列,而自动测序则采用激光扫描器同步扫描,计算机进行阅读和编辑
第二代 DNA 测序技术
第一代测序技术在分子生物学研究中发挥过重要的作用,如人类基因组计划(human genome project,HGP)主要基于第一代 DNA 测序技术。目前基于荧光标记和 Sanger 的双脱氧链终止法原理的荧光自动测序仪仍被广泛地应用。
随着人类基因组计划的完成,人们进入了后基因组时代,即功能基因组时代,第一代测序技术由于成本高、速度慢等原因已经不能满足深度测序和重复测序等大规模基因组测序的需求,这促使了第二代测序技术(高通量测序技术)。
第二代测序技术,主要包括:
- 罗氏 454 公司的 GS FLX 测序平台、
- Illumina 公司的 Solexa Genome Analyzer 测序平台
- ABI 公司的 SOLiD 测序平台
- Life Techologies 公司的离子肼测序
第二代测序技术最显著的特征是高通量,一次能对几十万到几百万条DNA分子进行序列测序,使得对一个物种的转录组测序或基因组深度测序变得方便易行。
第二代 DNA 测序技术原理
第二代测序技术将片段化的基因组 DNA 两侧连上接头,随后用不同的方法产生几百万个空间固定的 PCR 克隆阵列。每个克隆由单个文库片段的多个拷贝组成。然后进行引物杂交和酶延伸反应。由于所有的克隆都在同一平面上,这些反应就能够大规模平行进行,每个延伸反应所掺入的荧光标记的成像检测也能同时进行,从而获得测序数据。DNA 序列延伸和成像检测不断重复,最后经过计算机分析就可以获得完整的 DNA 序列信息。
454 测序流程
- DNA 文库制备:将待测 DNA 打断成 300 ~ 800 bp 长的小片段,末端修复后在片段两端加上不同的接头,变性处理回收单链 DNA。
- 体外 DNA 扩增: 454 系统采用的是乳化 PCR 的方法进行扩增。
- 富集固定:将含有 DNA 片段的磁珠富集起来,并将这些磁珠置于一种 PTP 平板中的特制小孔中,每个孔只能容纳一个磁珠。
- 测序:采用焦磷酸测序法,每个含有磁珠的小孔都可以测出其中一个片段的序列。
第二代 DNA 测序的局限性
第二代测序技术并不完美:
- 一是由于其在测序前要仍旧通过 PCR 手段对待测片段进行扩增,因此增加了测序的错误率
- 二是其测序结果比较短,更适合重测序,而不太适用于没有基因组序列的全新测序。
第三代测序技术(单分子测序,直接测序)
第三代测序技术主要有:
- Helicos公司的Heliscope 单分子测序仪
- Pacific Biosciences 公司的 SMRT 技术
- Oxford Nanopore Technologies 公司正在研究的纳米孔单分子技术
纳米孔单分子技术
- 使用外切酶从 ssDNA 的末端逐个切割形成单碱基
- 被切下来的单个碱基会落入纳米孔,并和纳米孔内的环糊精相互作用,短暂地影响流过纳米孔的电流强度,这种电流强度的变化幅度就成为每种碱基的特征。
- 利用不同碱基产生的电信号进行测序。
第三代测序技术特点
- 不再基于所用测序技术使用的边合成边测序的思想,减少合成过程中出现的差错;
- 测序步长达到 30 kb 以上。
第七章 DNA 文库的构建和目的基因的筛选
基因文库(Gene library):由某一生物的全部或部分基因的不同 DNA 片段的克隆所构成的群体, 称之为基因文库。一个完全的基因文库,应该能够保证从中筛选到目的基因。即 Genomic Library or Gene Bank
按照外源 DNA 的片段:
- 基因组 DNA 文库
- cDNA 文库
- 宏基因组文库
基因组 DNA 文库:指将某生物体的全部基因组 DNA 用限制性内切酶或机械力量切割成一定长度范围的 DNA 片段,与合适的载体体外重组并转化相应的宿主细胞获得的所有阳性菌落。包含基因的全部信息,如编码区,非编码区,内含子和外显子、启动子及调控序列
cDNA文库(complementary,DNA 互补DNA):由某一生物的特定器官或特定发育时期(时空特异性)细胞内的 mRNA 经反转录形成的 cDNA,它们所构成的重组 DNA 克隆群体,则称之为 cDNA 基因文库。
反映了基因表达谱,对研究基因表达,调控及基因互作非常有用。
DNA 文库构建的基本程序
- 提取研究对象基因组 DNA,制备合适大小的 DNA 片段,或提取组织或器官的 mRNA 并反转录成 cDNA;
- 酶切的 DNA 片段或 cDNA 与经特殊处理的载体连接形成重组 DNA;
- 重组 DNA 转化宿主细胞或体外包装后侵染受体菌;
- 阳性重组菌落或噬菌斑的选择。
高纯度大分子量基因组 DNA (High molecular weight DNA, HMW DNA)的提取
文库的代表性和随机性
文库中所有克隆所携带的 DNA 片段重新组合起来可以覆盖整个基因组,即可以从该文库中分离任何一段 DNA 。
- 采用酶切或随机切割的方法来消化染色体 DNA ,以保证克隆的随机性,保证每段 DNA 在文库中出现的频率均等;
- 增加文库总容量。
1976年 L.Clark,J.Carbon 提出了一个完全的基因文库所需克隆的计算公式$$n= \frac{\ln_{}{(1-p)}}{\ln_{}{(1-f)}}$$ $n$ : 一个完全基因文库所应包含的重组体克隆数
$p$ : 所期望的目的基因在基因文库中出现的几率
$f$ : 插入片段的平均大小与基因组 DNA 大小的比值
基因组 DNA 文库的构建
定义:指将某生物体的全部基因组 DNA 用限制性内切酶或机械力量切割成一定长度范围的 DNA 片段,与合适的载体体外重组并转化相应的宿主细胞获得的所有阳性菌落。
基因组 DNA 文库的构建程序:
- 载体的制备;
- 高纯度大分子量基因组 DNA (High molecular weight DNA, HMW DNA)的提取;
- HMW DNA 的部分酶切与脉冲电泳分级分离(PFGE size selection);
- 载体与外源片段的连接与转化或侵染宿主细胞;
- 重组克隆的挑取
- 文库的保存。
用于构建基因文库的载体:
- Plasmid,<10kb,亚基因组文库
- Phage,0 ~ 23 kb,cDNA文库
- Cosmid, ~ 45 kb,小基因组物种的基因组文库
- BAC, ~ 100kb,大基因组物种的基因组文库,物理图谱构建,基因组测序
- YAC, ~ 300kb ~ 1.2 Mb,大基因组物种的基因组文库,物理图谱构建,基因组测序
噬菌体改建的载体,利用了噬菌体的包装效率高和杂交筛选背景低的优点;经改造的人工染色体,其主要优点在于可容纳超过 100 kb 以上的外源片段。
粘粒克隆所需的克隆子数是 λ phage 的一半,如 λ 需 700000 时,cosmid 需 350000 个。YAC 可用于克隆 500 kb 以上,甚至几 Mb 的 DNA 片段,克隆子数就更少了。
亚基因组文库的构建
用基因组 DNA 的特定部分所构建的基因文库质粒 DNA,线粒体 DNA,特定限制性片段
在有杂交探针的情况下,可通过 Southern 杂交,先找出目标基因所在的限制性片段的大小,然后用相应大小的限制性 DNA 片段构建出基因文库,这样可减少筛选的压力。
例如,将基因组 DNA 用多种限制性内切酶切割,通过 Southern 杂交,发现目标基因在 8.3 kb SpeⅠ 片段上,则将SpeⅠ 切割的基因组 DNA 中的 ~ 8.3 kb 片段回收,用于构建 DNA 文库
基因组 DNA 文库克隆子的保存
- 影印滤膜保存法
- 文库在液体培养基中扩增保存
- 保存单个克隆子
文库在液体培养基中扩增保存
方法:从琼脂平板上挑取已长出的克隆子转入含适当的抗生素培养基中,混合的细菌生长数代后,其培养物于 -70℃ 储存(加终浓度为 25% 的甘油)。
缺点:因文库菌落生长的不均匀性而导致文库中某些特定的序列过多或过少。
保存单个克隆子
方法:从平板上挑选单个克隆子接种于合适的含抗生素的培养基中,菌体生长到一定浓度后,加入终浓度为 25% 的甘油,于 -70℃ 下保存。
缺点:需保存的克隆子数过多,工作量大。
cDNA 文库的构建
定义:是指将某种生物体基因组转录的全部 mRNA 经反转录产生的 cDNA 片段分别与克隆载体重组,储存于某种受体菌中,该群体就称该生物基因组的 cDNA 文库。
cDNA 文库的构建主要分四步:
- 细胞总 RNA 的提取和 mRNA 分离;
- 第一链 cDNA 合成;
- 第二链 cDNA 合成;
- 双链 cDNA 克隆进质粒或噬菌体载体并导入宿主中繁殖。
mRNA 的分离
从总 RNA 中富集 mRNA 是构建 cDNA 文库和其它应用所必需进行的步骤。通过降低 rRNA 和 tRNA 含量,可大大提高筛选到目标基因的可能性。
目前纯化 mRNA 的方法都是在固体支持物表面共价结合固定一段由脱氧胸腺嘧啶核苷组成的寡聚核苷酸 [oligo(dT)] 链,由它与 mRNA 的 Poly(A) 尾巴杂交,从而吸附固定住 mRNA,进而将 mRNA 从其它组分中分离出来的。
(1)mRNA 的含量:取决于细胞类型和细胞的生理状态。
(2)mRNA 完整性的检测:
- mRNA 分子的大小:哺乳动物 mRNA 长度为 500 ~ 8000 bp,大部分 mRNA 位于 1.5 ~ 2.0 kb 之间。
- 总 mRNA 指导合成 cDNA 第一链长分子的能力。
- 指导合成高分子量蛋白质的能力,指导合成目的多肽的能力。
(3)mRNA 在细胞中的丰度
- 高丰度 mRNA:目的 mRNA 在细胞中的含量占细胞质总 mRNA 量的 50 ~ 90%,如珠蛋白,免疫球蛋白,卵清蛋白,该类 mRNA 在合成和克隆 cDNA 之前不需进一步纯化特定 mRNA。
- 低丰度 mRNA:目的 mRNA 在细胞中的含量占细胞质总 mRNA 量的 0.5% 以下。
(4)对 mRNA 进行分级分离
- 通过琼脂糖凝胶电泳分离大小不同的 mRNA 分子,该方法的分离效果最好,但从凝胶中回收的得率较低。
- 蔗糖梯度离心:加入破坏 RNA 二级结构的变性剂如氢氧化甲基汞等,再进行蔗糖梯度离心以分离不同分子量的 mRNA。
cDNA 的分级分离:mRNA 通过反转录形成 cDNA,在插入到克隆载体前,通过琼脂糖凝胶电泳,将不同大小的 cDNA 分子分离开来。优点:
- 避免了分离过程中 mRNA 被污染的 RNA 酶降解
- 增加了获得全长 cDNA 克隆的概率
- 获得更准确的分级分离效果(分子量)
第一链 cDNA 的合成
oligo(dT) 引导的 DNA 合成法:利用真核 mRNA 分子所具有的 poly(A) 尾巴的特性,加入 12 ~ 20 个脱氧胸腺嘧啶核苷组成的 oligo(dT) 短片段,由反转录酶合成 cDNA 的第一链。
第二链 cDNA 的合成
cDNA 第二链的合成就是将上一步形成的 mRNA-cDNA 杂合双链变成互补双链 cDNA 的过程。
cDNA 第二链的合成的方法大致 4 种:
- 自身引导合成法
- 置换合成法
- 引导合成法
- 引物-衔接头合成法。
(一)自身引导合成法
获得的单链 cDNA 3’ 端会形成发夹结构的能力,以此作为第二链合成的引物,在大肠杆菌聚合酶ⅠKlenow 或反转录酶的作用下,合成 cDNA 的第二链。再利用 S1 核酸酶将连接处(仅该位点处为单链结构)切断形成平端结构可以进行连接。
缺点:在以 S1 核酸酶切割 cDNA 的发夹状结构时,会导致对应于 mRNA 5’ 端的地方的序列出现缺失和重排。S1 核酸酶的纯度不够时,会偶尔破坏合成的双链 cDNA 分子。
(2)置换合成法
以第一链合成产物 cDNA:mRNA 杂交体作为切口平移的模板,RNA 酶 H 在杂交体的 mRNA 链上造成切口和缺口,产生一系列 RNA 引物,在大肠杆菌 DNA 聚合酶Ⅰ的作用下合成 cDNA 的第二链。
优点:
- 合成 cDNA 的效率高
- 直接利用第一链的反应产物,不需纯化
- 避免使用 S1 核酸酶来切割双链 cDNA
(3)引导合成法
首先是制备带有 Poly(dT) 的载体片段Ⅰ和一端带有 Poly(dG) 的片段Ⅱ,并用片段Ⅰ来代替 Oligo(dT) 进行 cDNA 第一链的合成,在第一链 cDNA 合成后直接采用末端转移酶(TdT)在第一链 cDNA 的 3’-端加上一段 Poly(dC) 的尾巴,同时进行酶切创造出另一端的粘端,与片段Ⅱ一起形成环化体,这种环化了的杂合双链在 RNA 酶 H、大肠杆菌 DNA 聚合酶Ⅰ和 DNA 连接酶的作用下合成与载体联系在一起的双链 cDNA 。
(4)引物-衔接头法:引导合成法改进而来
第一链合成后直接采用末端转移酶(TdT)在第一链 cDNA 的 3’-端加上一段 Poly(dC) 的尾巴,然后用一段带接头序列的 Poly(dG) 短核苷酸链作引物合成互补的 cDNA 链,接头序列可以是适用于 PCR 扩增的特异序列或用于方便克隆的酶切位点的序列。这一方法目前已经发展成 PCR 法构建 cDNA 文库的常用方法。
双链 cDNA 克隆进质粒或噬菌体载体并导入宿主中繁殖
cDNA 末端的处理:
- 由于大片段的平末端连接效率非常低,因此为了避免用平末端与载体连接,对双链 cDNA 的末端进行加工是十分必要的
- 方法:添加特异性核酸接头以形成适合于克隆的黏性末端
- 末端转移酶,可以向 cDNA 末端加上与克隆载体末端互补的尾部
双链 cDNA 的克隆,双链平头的 cDNA 通常可以使用下列三种方法克隆入载体中:
- 平头末端直接与载体连接,但连接效率低
- 平头两端分别接同聚物尾,最好是 AT 同聚物尾,这样重组分子可通过加热局部变性和 S1 核酸酶处理回收插入片段
- 加装人工接头引入酶切口,以便插入片段回收
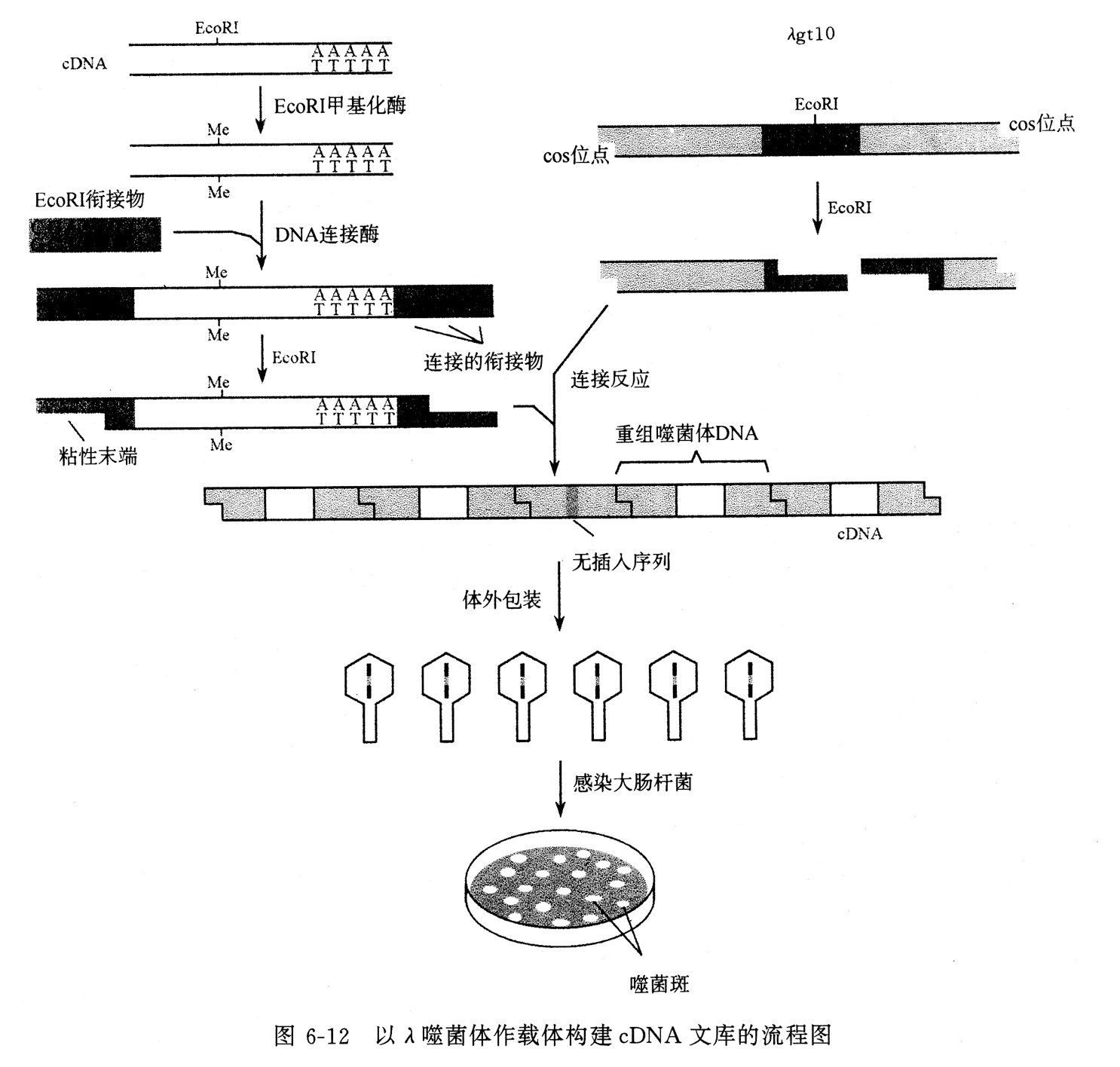
基因组 DNA 文库与 cDNA 文库的比较
相对于 cDNA 文库,基因组文库的优点:
- cDNA 克隆只能反映 mRNA 的分子结构,没有包括基因组的间隔序列,并且受细胞来源或发育时期的影响。
- cDNA 文库中,不同克隆的分布状态总是反映 mRNA 的分布状态,即:高丰度 mRNA 的 cDNA 克隆,所占比例较高,分离基因容易;低丰度 mRNA 的 cDNA 克隆,所占比例较低,分离基因困难;
- 从 cDNA 克隆中,不能克隆到基因组 DNA 中的非转录区段序列,不能用于研究基因编码区外侧调控序列的结构与功能。
cDNA 文库的主要优点:
- cDNA 文库以 mRNA 为材料,特别适用于某些 RNA 病毒等的基因组结构研究及有关基因的克隆分离。
- cDNA 文库的筛选比较简单易行。
- 每一个 cDNA 文库都含有 mRNA 序列,这样在目的基因的选择中出现假阳性的概率就会比较低,因此阳性杂交信号一般都是有意义的,由此选择出来的阳性克隆将会含有目的基因。
- cDNA 克隆还可用于真核细胞 mRNA 的结构和功能研究。
基因克隆的筛选策略
- 表型筛选法
- 杂交筛选和 PCR 筛选
- 免疫筛选
- 酵母双杂交系统筛选
酵母双杂交系统
酵母双杂交系统筛选:用于分离与某一已知蛋白发生相互作用的蛋白质基因
GAL4 蛋白:酵母半乳糖苷酶基因 gal 的转录激活因子,该蛋白结合在 gal 基因上游激活区 (UAS) 可启动 gal 基因的转录
GAL4 蛋白可分为两个区域:
- DNA-BD:DNA 结合域,N-末端 1 ~ 147 aa(氨基酸)
- AD:转录激活域,C-末端 768 ~ 881 aa
这两个结合域将它们分开时仍分别具有功能,但不能激活转录,只有当被分开的两者通过适当的途径在空间上较为接近时,才能重新呈现完整的转录因子活性,并可激活上游激活序列(upstream activating sequence, UAS)的下游启动子,使启动子下游基因得到转录。
酵母双杂交系统筛选原理
- 将编码 DNA-BD 的基因与已知蛋白质 Bait protein 的基因构建在同一个表达载体上,在酵母中表达两者的融合蛋白 BD-Bait protein。
- 将编码 AD 的基因和 cDNA 文库的基因构建在 AD-LIBRARY 表达载体上。
- 同时将上述两种载体转化改造后的酵母,如两种载体所表达的融合蛋白不相互作用时,这种改造后的酵母细胞的基因组中既不能产生 GAL4,又不能合成 LEU、TRP、HIS、LacZ,因此,酵母在缺乏这些营养的培养基上无法正常生长。当上述两种载体所表达的融合蛋白能够相互作用时,功能重建的反式作用因子能够激活酵母基因组中的报告基因 HIS、LacZ 等,从而通过功能互补和显色反应筛选到阳性菌落。
酵母双杂交系统的优点
- 作用信号是在融合基因表达后,在细胞内重建转录因子的作用而给出的,省去了纯化蛋白质的繁琐步骤。
- 检测在活细胞内进行,可以在一定程度上代表细胞内的真实情况。
- 检测的结果可以是基因表达产物的积累效应,因而可检测存在于蛋白质之间的微弱的或暂时的相互作用。
- 酵母双杂交系统可采用不同组织、器官、细胞类型和分化时期材料构建 cDNA 文库,能分析细胞质、细胞核及膜结合蛋白等多种不同亚细胞部位及功能的蛋白。
酵母双杂交系统局限性和存在的问题
- 双杂交系统分析蛋白间的相互作用定位于细胞核内,而许多蛋白间的相互作用依赖于翻译后加工如糖基化、二硫键形成等,这些反应在核内无法进行这限制了某些细胞外蛋白和细胞膜受体蛋白等的研究。
- 酵母双杂交系统的一个重要的问题是"假阳性"。由于某些蛋白本身具有激活转录功能或在酵母中表达时发挥转录激活作用,使 DNA 结合结构域杂交蛋白在无特异激活结构域的情况下可激活转录。另外某些蛋白表面含有对多种蛋白质的低亲和力区域,能与其他蛋白形成稳定的复合物,从而引起报告基因的表达,产生"假阳性"结果。
第八章 植物基因工程
植物基因工程概念
植物基因工程是以植物为受体,采用基因克隆、遗传转化以及细胞、组织培养技术将外源基因转移并整合到受体植物的基因组中,并使其在后代植株中得以正确表达和稳定遗传,从而使受体获得新性状的技术体系。
转基因植物快速发展的因素
- 植物单个细胞具有发育成完整个体的“细胞全能性”;
- 植物是人类食物和能量的主要来源
- 许多高等植物具有自花授粉或自交能力,易于得到纯合的转基因个体
- 具有丰富的突变体资源,为植物基因的分离提供良好的条件。
植物转基因的基本技术路线
- 获得目的基因,克隆到载体上扩增
- 受体细胞培养:如植物愈伤组织等
- 目的基因导入受体细胞
- 培养转化细胞
- 筛选、培植、鉴定
转基因的受体系统
- 组织受体系统(如:受伤的细胞容易受到病毒或质粒的感染,并形成愈伤组织,可诱导分化出完整植株)
- 原生质体系统(便于体外细胞和遗传操作)
- 生殖细胞受体系统(单倍体培养诱导愈伤组织、转化;利用花粉和卵细胞的受精过程转化)
- 叶绿体转化系统(能直接表达原核基因)
外源基因导入植物受体的方法
- 根癌农杆菌介导法
- 基因枪法
- 原生质体介导法
根癌农杆菌介导法
一种土壤细菌感染植物伤口造成植物的肿瘤(冠瘿)
植物受伤处的分泌物(糖、氨基酸和酚类物质等)诱导农杆菌的附着,农杆菌中的 T-DNA 插入植物基因组,T-DNA 利用植物的酶系统进行转录和翻译,其表达产物致使植物致瘤。
Ti 质粒:
- T-DNA 区,转移到植物基因组的一段 DNA
- 毒性区(vir区):激活 T-DNA 转移,使植物致瘤
- Ori区:质粒复制起点
- Con区:调控 Ti 质粒在根癌农杆菌间的转移
Ti 质粒分为:农杆碱型、胭脂碱型、章鱼碱型
T-DNA 的转化需要 Vir 区基因的表达和左右边界(LB、RB)的存在
土壤农杆菌-植物 DNA 转移体系步骤:
- 农杆菌对受体的识别;
- 农杆菌附着到植物受体细胞;
- Ti 质粒毒性区基因被激活表达;
- 类似接合孔复合体的合成和装配;
- T-DNA 的切割、加工和转运;
- T-DNA 整合到植物染色体上
T-DNA 整合到植物染色体上:
- 首先在植物靶 DNA 上出现一个缺刻(nick)
- 随着 DNA 解链,宿主细胞的 5’-3’ 外切酶活性使缺刻扩大成裂口(gap)
- T-DNA 侵入裂口,末端与靶 DNA 单链上的少数核苷酸配对形成异源二倍体;
- T-DNA 悬挂在外侧的末端被切割除去,T-DNA 与靶 DNA 末端相连;
- 以整合后的 T-DNA 链为模板合成 T-DNA 的第二条链,完成整合过程。
Ti 质粒的改造
改造原因:
- Ti 质粒无单一的限制性内切酶位点,不能通过体外 DNA 重组技术直接向野生型 Ti 质粒导入外源基因;
- Ti 质粒的分子量太大,给 DNA 的重组以及基因转化等操作带来困难,Ti 质粒上还存在一些对于 T-DNA 转移不起任何作用的基因。
- 野生的 Ti 质粒带有致瘤基因,感染植物后,引起冠瘿瘤,阻碍细胞的分化和植株的再生;
- Ti 质粒本身无合适的选择基因;
- Ti 质粒不能在大肠杆菌中复制,即使得到重组质粒,也只能在农杆菌中进行扩增。
Ti 质粒衍生的克隆转化载体必须具备以下的结构特点:
- 具有选择标记基因
- DNA 复制起始位点
- T-DNA 右边缘序列
- 单克隆位点
- vir 区域,利用双元载体系统和共整合载体系统解决。
共整合载体系统(一元载体系统)
- Ti 质粒进行卸甲处理
- 将目的基因构建于中间载体上
- 在卸甲载体 T-DNA 区插入一段与中间载体同源的质粒序列。
- 然后将中间载体转移到含有卸甲载体的根癌农杆菌中,中间载体通过同源重组整合到卸甲载体的 T-DNA 区,并与卸甲 Ti 质粒一起复制。
- 根据中间载体上所携带的抗性基因进行抗性筛选,获得遗传重组根癌农杆菌菌株。
- 使用这种菌株去侵染植物组织细胞,就可以获得含有目的基因的转基因植物。
双元载体系统:Vir 基因不必与 T-DNA 位于同一质粒上,只需处于同一细菌中
叶盘转化法
基因枪法
基因枪法原理:借助高速运动的金属微粒(金粒或钨粒)将质粒 DNA 引入植物细胞内。首先将质粒 DNA 附着在金属微粒子上,然后借助高压驱动,将附有 DNA 的微粒射入靶细胞内。
决定基因枪使用成功的因素:
- 第一因素是动力系统。根据动力系统,基因枪分为三大类:
- 第一类是以火药爆炸力作为动力加速微弹;
- 第二类是以电弧放电蒸发浪作为动力;
- 第三类是以高压气体作为动力。
- 第二因素是微弹的制备过程。用的微载体有两类:一是钨粉,另一个是金粉,直径一般为 0.6 ~ 4 μm。常用的将 DNA 包被到微载体上所用的沉淀试剂有亚精胺、氯化钙、乙醇、聚乙二醇、异丙醇等。
- 第三个因素是受体材料的选择。
原生质体介导法
(一)PEG 介导原生质体转化
原理:在 PEG 和 Ca2+ 的存在下,原生质体的细胞膜会发生一系列结构上的瞬时变化,使得 DNA 通过细胞膜进入细胞内,进
而与植物基因组 DNA 整合。
机制:推测在二价阳离子存在条件下,PEG 使 DNA 沉淀在细胞膜上,细胞通过内吞作用,将质粒 DNA 吞进细胞。
转化过程:原生质体的制备 → 转化 → 培养 → 筛选抗性愈伤组织 → 再生
转化受体及特点:受体原生质体,已成功转化水稻、小麦等,技术性强,程序复杂,转化频率较低,转基因植株变异频率高。
(二)脂质体介导法
脂质体:是一种人工膜。在水中磷脂分子亲水头部插入水中,疏水尾部伸向空气,搅动后形成双层脂分子的球形脂质体,直径 25 ~ 1000 nm不等。
用脂质体包裹一些 DNA 就成了一种人工模拟的原生质体,与植物原生质体共保温后脂质体与原生质体膜结构之间发生相互作用,而后通过细胞的内吞作用而将外源 DNA 导入植物的原生质体。
(三)电激法
在高压电场的瞬时作用下,细胞膜可以出现可逆的微小孔洞,外源 DNA 可以通过这些孔洞进入细胞内。可用于真核、原核细胞的转染
优点:简单、重复性好、转移效率高
缺点:对细胞有损伤
转化受体及特点:受体原生质体或部分消解的愈伤组织,已成功转化水稻、玉米、小麦、烟草、马铃薯等,转化频率较高
(四)显微注射法
DNA 损失少
无需选择标记基因整合效率高
(五)激光微束介导法
利用激光微束(0.3 ~ 0.5 uM)射击靶细胞,引起可逆性穿孔,从而直接导入外源 DNA。首先在动物与人的细胞中获得成功,80 年代后期用于植物。
技术原理:激光照射系统产生高能量激光脉冲,照射经高渗处理的材料,产生穿孔后,外源 DNA 向靶细胞渗入。然后培养靶组织,产生转化植株。
优点:可瞄准靶细胞定位导入外源 DNA,与其他方法比较对靶细胞损伤小、恢复快。单、双子叶植物均可,受体细胞可以是花粉、单细胞或组织、器官。
缺点:需要相应昂贵的设备,穿刺处理速度较慢。
植物转基因方法比较
根癌农杆菌介导法优点:可以在原生质体、细胞团、组织器官或整株等多级水平上进行,方法成熟可靠,简便易行,周期短,转化率高
根癌农杆菌介导法缺点:转化双子叶植物为主。大多数单子叶植物和裸子植物对农杆菌的侵入不敏感,限制了该法在禾谷类作物中的应用。
DNA 直接转化法优点:无宿主限制,适用于各种单、双子叶植物,操作简单
DNA 直接转化法缺点:转化效率低,需要专门设备(电激仪、显微操作仪或基因枪),多数需要原生质体与愈伤组织,周期太长
高等植物的基因表达系统
- 外源基因的四环素诱导系统
- 外源基因的乙醇诱导系统
- 外源基因的地塞米松诱导系统
- 外源基因的类固醇诱导系统
外源基因的四环素诱导系统
四环素
四环素阻碍蛋白基因(Tet repressor,Tet R)
四环素操纵基因(Tet resistance operon,TetO)
四环素诱导型目的基因(报道基因)
转基因植物的筛选与检测
选择性基因:
- 新霉素抗性基因:卡那霉素、新霉素、G418
- 庆大霉素抗性基因:庆大霉素
- 潮霉素磷酸转移酶基因:潮霉素
- 膦丝菌素乙酰转移酶基因:膦丝菌素
报告基因是指一类在细胞、组织、器官或个体处于特定情况下会表达并使得他们产生易于检测、且实验材料原本不会产生的性状的基因。↩
作为报告基因,在遗传选择和筛选检测方面须具有以下条件:
- 已被克隆和全序列已测定;
- 表达产物在受体细胞中本不存在,即无背景,在被转染的细胞中无相似的内源性表达产物,且不损坏受体细胞。
- 其表达产物能进行定量测定。
报告基因主要类型:
- 氯霉素乙酰转移酶基因(cat)
- β-D-葡萄糖酸苷酶基因(gus)
- 荧光素酶基因(luc)
- 绿色荧光蛋白基因(gfp)
转基因植物的 GUS 检测原理:β-D-葡萄糖酸苷酶基因(Gus),催化 β-葡萄糖苷酯类物质水解。其中很多水解产物具有发色团或形成荧光物质。
例:组织化学法测定 Gus 活性,作用底物为 X-gluc,产物是一种不可溶的 5,5-二溴,4,4-二氯靛蓝;
转基因植物的 GFP 检测
转基因植物产品的检测,目前已采用的检测方法:
- 核酸水平,DNA:PCR、Southern 杂交;RNA:RT-PCR、Nouthern 杂交
- 蛋白质水平,Western杂交
转基因植物的应用
- 抗除草剂的转基因植物(最早进入田间生产,目前载培面积最大),如 Monsanto 的抗除草剂草甘膦大豆
- 抗虫转基因植物,如Syngenta的Bt :176抗虫玉米
- 抗病转基因植物(抗病毒,抗真菌,抗细菌)
- 抗环境胁迫转基因作物(温度、 水分、化学物)
- 植物发育调节基因工程(控制果实成熟, 雄性不有,改良植物品质)
- 医药领域中的转基因植物(利用植物作为生物反应器,生产药用蛋白、食用疫苗等)
第九章 动物基因工程
转基因动物(transgenic animal):分为遗传性与非遗传性
- 遗传性(转基因动物个体):指用 DNA 重组技术将人们所需要的目的基因导入动物的受精卵或早期胚胎内,使外源目的基因随细胞的分裂而增殖并在体内表达,且能稳定地遗传给后代的动物。
- 非遗传性(动物细胞工程):指用 DNA 重组技术将人们所需要的目的基因导入动物受体细胞,使外源目的基因随细胞的分裂而增殖并在体内表达,仅在当代表现,不遗传给子代。
哺乳动物细胞表达系统优缺点
优点:表达的蛋白与天然蛋白在结构、糖基化、磷酸化、寡聚体类型和方式上几乎相同,且能正确组装成多亚基蛋白。如 EPO(促红细胞生成素)
EPO(促红细胞生成素)用原核系统进行表达——缺少可用以进行糖基化的结构和机制,酵母、昆虫或植物表达的产品可以糖基化,但这类细胞的糖基化酶不同,因而表达产物的寡糖链末端多为甘露糖、N-羟基乙酰神经氨酸(GlcNAc)和半乳糖,易被肝细胞、巨噬细胞表面的受体识别而清除。因此此类产品可能对人有免疫原性。
缺点:
- 哺乳动物细胞的表达水平低;
- 获得高表达细胞株所需的时间长;
- 细胞大规模培养的成本高;
- 哺乳动物细胞生产的蛋白质类药物的成本较高;
哺乳动物的受体系统
(一)正常的哺乳类动物细胞具有四大生物学特征:
- 锚地依赖性:细胞必须附在固体上或固定的表面才能生长分裂
- 血清依赖性:细胞必须具有生长因子才能生长
- 接触抑制性:细胞与细胞接触后,生长便受到抑制
- 形态依赖性:细胞扁平状,并有长纤维网状结构
上述特征使得正常的哺乳动物细胞在体外培养中,一般只能存活 50 代且在培养皿上以平面的形式生长,即单层细胞生长。有时,正常细胞会改变某些特征而越过生理临界点,继续增殖并无限制分裂,这种状态称为细胞系形成,此时的细胞成为细胞系
(二)高效表达外源基因受体细胞应具备下列条件:
- 细胞系特征,丧失细胞接触抑制性和锚地依赖性特征,便于大规模培养
- 合适的标记,便于转化株的筛选和维持
- 遗传稳定性,外源基因多次传代后不至于丢失,易于长期保存
- 生长快且齐,分裂周期短,生长均一,便于控制
- 安全性能好,不合成分泌致病物质,不致癌
(三)高等哺乳动物受体细胞的类型
根据受体细胞表达的重组蛋白的稳定性和糖基化类型不同,按照要表达的目的蛋白选择最佳的受体细胞,现今高等哺乳动物受体细胞类型主要由以下 5种:
- 中国仓鼠卵巢细胞(CHO)
- 绿猴肾细胞(CV-1)
- 地鼠幼鼠肾细胞(BHK)
- 小鼠乳腺肿瘤细胞(C127)
- 西班牙犬肾细胞(MDCK细胞)
(1)中国仓鼠卵巢细胞(CHO),迄今为止,用于医疗用品(药物、抗体、诊断试剂)大规模生产的高等哺乳动物受体细胞主要是中国仓鼠卵巢细胞(CHO)
- 遗传背景清楚,生理代谢稳定
- 与人的亲缘关系接近,外源蛋白修饰准确
- 基因转移和载体表达系统完善
- 耐受剪切力,便于大规模培养
- 被美国 FDA 确认为安全的基因工程受体细胞(GRAS)
(2)1981 年,Gluzman 用编码野生型 T 抗原但复制起点缺失的 SV40,转化允许 SV40 裂解性生长的非洲绿猴肾细胞 CV-1,获得了三个细胞系:COS1、COS3、COS7。这三个细胞系均含有 T 抗原,保留完全的允许 SV40 裂解性生长的能力(被广泛地用于瞬时表达系统)。
T 抗原作用:当表达产生的 T 抗原结合到 SV40 的复制点的 DNA 调控区,病毒 DNA 的复制即被启动。
(3)BHK-21:1961 年从地鼠幼鼠的肾脏分离而来成果:用它表达的重组凝血因子Ⅷ已获准投放市场。
(4)C127 细胞:来自 RⅢ 小鼠乳腺肿瘤细胞,特别适用于带有牛乳头瘤病毒(BPV)载体的转染:用 C127 细胞生产的重组人生长激素(hGH)已获准投放市场,用于治疗生长激素缺乏症。
(5)MDCK 细胞:1958 年从成年雌性的西班牙长耳狗的肾脏分离获得的,是贴壁生长的上皮样细胞:有报道用该细胞结合人巨细胞病毒早早期启动子可高效表达分泌蛋白,表达量占细胞分泌蛋白质总量的 15% ~ 20%。
高等哺乳动物受体细胞的遗传标记

含有胸腺嘧啶核苷激酶(TK)编码基因缺陷的(tk-)受体细胞,不能在含有次黄嘌呤、氨基喋呤、胸腺嘧啶核苷的培养基上(HAT 培养基)生长,载体上的标记基因 tk 能与之遗传互补
含有次黄嘌呤磷酸核糖转移酶(HPRT)编码基因缺陷的(hprt-)受体细胞,不能在含有次黄嘌呤、氨基喋呤、胸腺嘧啶核苷的培养基上(HAT培养基)生长,载体上的标记基因 hprt 与之遗传互补
哺乳动物细胞载体系统
哺乳动物细胞表达载体应具备的条件:
- 基本的元件:增强子、终止信号和 poly(A) 信号、多克隆位点、剪接信号、筛选标记等;
- 易于扩增至足够数量,能够在哺乳动物细胞内稳定存在、不丢失:哺乳动物细胞无天然质粒,需具有原核和真核复制起点;
- 能够在哺乳动物细胞内高效表达的强启动子:一般细胞不过量表达蛋白。
质粒型载体
(一)通用型表达载体
通用型表达载体拥有真核细胞表达载体共同结构特征,可使携带的目的基因高效表达,而且一般无物种或细胞类型的特异性,是研究基因功能的有力工具。如 pEGFP-N1 等。
pEGFP-N1 表达载体的特点:
- 从结构上看,载体中的 pUC ori,保证该载体在大肠杆菌中具有很强的复制能力;
- 含有高效且功能强大的启动子 PSV40 和 PCMV,可以使目的基因在细胞中稳定表达;
- 具有多克隆位点(MCS),便于目的基因的插入;
- 具有 SV40 ori,使该载体在任何表达 SV40 T 抗原的真核细胞内进行复制;
- 含新霉素抗性基因表达盒,由 SV40 早期启动子、卡那霉素抗性基因(neor/kanr) 以及单纯疱疹病毒胸苷激酶基因 HSV-TK 的聚腺嘌呤信号组成,可以采用 G418 来筛选。
- 具有 EGFP 报告基因。EGFP 是一种优化的突变型 GFP,产生的荧光比普通 GFP 强 35 倍,大大增强了其报告基因的敏感度。EGFP 的 N 及 C 端均可融合,并不影响其发光。
这些特殊的结构可以实现目的基因在靶细胞内的稳定表达和鉴定。
(二)组织特异性表达载体
组织特异性表达载体是利用某种组织特殊的启动子构建真核表达载体,驱动目的基因在特定组织中表达。常用的高等哺乳动物组织特异性表达载体有乳腺组织特异性表达载体、脂肪组织特异性表达载体、肌肉组织特异性表达载体、神经组织特异性表达载体、前列腺组织特异性表达载体等
pBC1 是山羊乳腺特异性质粒表达载体,具有以下特点:
- 含有一个可以保证重组质粒在原核细胞内大量复制的复制原点(来自质粒 pBR322);
- 具有选择性标记氨苄青霉素抗性基因(ampicillin)
- 具有山羊乳腺特异性表达的 β-酪蛋白基因启动子(Pβ-casein),目的基因插入到外显子 2 和 7 之间的 XhoⅠ 多克隆位点上,其后
为 β-酪蛋白基因的 3’ 调控区,保证转录的有效终止。 - 启动子上游有两个 β-肌球蛋白(β-globin)基因的绝缘子序列(β-globin insulator),能够防止随机整合外源基因插入位点的位置效应,保证目的基因的高效表达。
- 该载体还可以通过在 SalⅠ 和 NotⅠ位点插入靶基因的两侧同源臂,制备成同源重组转基因结构,实现基因敲除(knock-out)与敲入(knock-in)。
利用该转基因结构制备的转基因动物乳腺中外源基因的表达水平可达到 60 mg/L。
病毒类载体
- 人腺病毒 DNA
- 猴空泡病毒 DNA(SV40)
- 人乳多瘤病毒 DNA(BKV)
- 人牛痘病毒 DNA
腺病毒的基因组 DNA
腺病毒基因组 DNA 全长 36 kb,其包装上限为原基因组的 105%,DNA 两端各有一个反向重复序列(ITR);E1 ~ E4为早期基因,与病毒基因组表达调控有关,其中 E3 编码晚期基因的调控因子,L1 ~ L5 为编码病毒包装蛋白的晚期的复制及晚期基因的基因;IVa2 和 VA 均为病毒 RNA 聚合酶的亚基编码基因。E3 区缺失只会影响病毒颗粒的成熟,不影响基因组的复制功能,因而在构建载体时往往除去这个 2.2 kb的片段,使得载体的装载量提高到 4 kb以上。
腺病毒 DNA 载体的特点:
- 基因重排低,外源基因与病毒 DNA 重组后能稳定复制几个周期
- 安全性能好,不整合人的染色体 DNA,不会导致恶性肿瘤
- 宿主范围广,对受体细胞是否处于分裂期要求不严格
- 使用效果好,外源基因在载体上容易高效表达
定向打靶载体
基因打靶(genetargeting)是一种定向改变生物活体遗传信息的操作技术。该技术是在胚胎干细胞(embryonic stem cell, ESC)与同源重
组(homologous recombination, HR)基础上建立起来的。基因打靶通过对生物活体遗传信息的定向修饰包括基因灭活、点突变引入、缺
失突变、外源基因定位引入、染色体组大片段删除等,并使修饰后的遗传信息在生物活体内遗传,表达突变的性状,从而研究基因功能、提供相关的疾病治疗和建立新药饰选评价模型等。2007 年度诺贝尔生理学或医学奖授予了美国犹他大学医学院的 Capecchi、英国卡迪夫大学医学院的 Evans 和北卡罗莱纳大学医学院的 Smithies 等三人以表彰他们在小鼠中的“基因打靶”技术的应用。
基因打靶技术主要四个步骤:
- 基因打靶载体的构建。把目的基因和调控序列等与内源靶序列同源的序列都重组到带标记基因的载体上。
- 打靶载体的导入受体细胞内;
- 同源重组子的筛选,用选择性培养基筛选打靶击中的重组阳性细胞;
- 将重组阳性细胞转入动物胚胎,产生转基因动物,并进行检测。
(1)敲降载体
敲降载体的作用是使受体细胞特定的靶基因 mRNA 发生降解,从而使靶基因的表达水平大幅降低。
敲降载体的构建是基于 RNAi 技术原理进行的。RNAi 的优点是作用特异性强,敲降靶基因效率高,相对基因敲除载体操作简单,筛选细胞周期短。
由于 RNAi 识是在转录后水平上降低基因表达,而不是从基因组中剔除掉基因,有时也会因背景不干净导致表型难以分析。
以荧光素酶报告基因为例说明其检测原理。首先构建一个真核表达载体,在荧光素酶或 GFP 报告基因的翻译终止密码子后插入目的基因片段。将构建后的载体和 shRNA 共转染 293 T 细胞,在 293 T 细胞内就会转录出荧光素酶基因和目的基因的融合 mRNA,如果 shRNA 在目的基因上无靶点,不会影响荧光素酶基因的翻译,在添加荧光素酶化学发光底物情况下,底物就会被分解并产生一定波长光,通过检测器可检测荧光;反之,若 shRNA 在目的基因上有靶点,融合 mRNA 被降解,荧光素酶基因不能翻译,就不会产生荧光。通过该系统可以快速、准确的验证 RNAi 干扰靶点。
(2)敲除载体
敲除载体是在 DNA 水平对受体细胞内靶基因实施编辑与修饰,对基因功能研究、转基因动物疾病模型建立和畜禽性状改良等方面有着广泛应用价值。
敲除载体主要作用是破坏靶细胞中特定基因表达结构,使其不能表达,具有位点特异性。为了实现此目的,可通过同源重组来实现,随着基因敲除技术的发展产生了基因敲除效率更高的锌指核酸酶、TALEN、CRISPR/Cas9 和 Cre-LoxP等技术。
CRISPR/Cas 系统概述
CRISPR/Cas 系统是很多细菌和大部分古细菌的天然免疫系统,通过对入侵的病毒和核酸进行特异性的识别,利用 Cas 蛋白进行切割,从而达到对自身的免疫。
CRISPR 结构:CRISPR 是一种特殊的 DNA 重复序列家族,广泛分布于细菌和古细菌基因组中。CRISPR 位点通常由短的高度保守的重复序列(repeats)组成,重复序列的长度通常为 21 ~ 48 bp,重复序列之间被 26 ~ 72 bp 间隔序列(spacer)隔开。CIRSPR 通过这些间隔序列(spacer)与靶基因进行识别。
Cas 家族:Cas(CRISPR associated)存在于 CRISPR 位点附近,是一种双链 DNA 核酸酶,能在 guide RNA 引导下对靶位点进行切割。它与 fokⅠ 酶功能类似,但是它并不需要形成二聚体才能发挥作用。
CRISPR/Cas9 系统作用机制
- CRISPR 基因座的表达(包括转录和转录后的成熟加工)
- 当该噬菌体再次入侵细菌时,CRISPR 簇首先转录为长的 crRNA 前体,然后逐步加工成小的成熟的 crRNA。
- CRISPR/Cas 系统活性的发挥或外源遗传物质的干扰
- crRNA 结合相关的 Cas 蛋白后,形成 crRNA-Cas 蛋白复合体,通过碱基互补配对精确地与目标 DNA 相结合,随后 Cas 蛋白对目标 DNA 进行断裂和降解。
- Cas9 是一种核酸内切酶,其具有:RuvC 和 HNH 两个内切酶活性中心
- Jinek 等发现 Cas9 在细菌和试管里对双链 DNA 具有强烈的切割能力,但是这种切割能力需要的 crRNA 和 tracrRNA 介导。
- Jinek 等创造性的把 crRNA 和部分 tracrRNA 融合成一条嵌合的 RNA 链,使其同时具有 crRNA 和 tracrRNA 的特性,随后的切割实验表明,这条嵌合的 RNA 同样可以引导 Cas9 切割目标 DNA。现在普遍使用的都是 crRNA 和全长 tracrRNA 融合体,简称 sgRNA (single guide RNA)
CRISPR/Cas9 脱靶效应
- 2013 年,研究者们发表多篇文章介绍 CRISPR/Cas9 系统,但 CRISPR/Cas9 目前仍存在一个很严重且无法避免的问题,即潜在的脱靶效应不可消除。
- Cas9 蛋白不具特异性
- CRISPR/Cas9 与靶位点识别的特异性主要依赖于 gRNA 与靠近 PAM(5’-NGG-3’)处 10 ~ 12 bp 碱基的配对,而其余远离 PAM 处 8 ~ 10 bp 碱基的错配对靶位点的识别影响不大。
降低脱靶效应:添加同源臂(homology arm,HR)
制备转基因动物的方法
转基因动物研究的核心技术是成功地把目的基因转入动物早期胚胎细胞中。目前,研究制备转基因动物的主要方法有:
- 显微注射法
- 胚胎干细胞法
- 反转录病毒感染法
- 精子载体导入法
- 体细胞核移植法
显微注射法
受精卵能够分化出各种细胞、组织,形成一个完整的个体,所以把受精卵的分化潜能称为全能性。随着分化发育的进程,转基因会分布到各种组织细胞中去,包括生殖细胞,因此,转基因是可以遗传的。
受精卵显微注射制备转基因动物的优点:
- 转移率高
- 实验周期相对较短
- 导入过程直观
- 外源基因的大小不受限制
- 没有化学试剂等对细胞的毒性作用
受精卵显微注射制备转基因动物的缺点:
- 设备昂贵;
- 技术难度高;
- 胚胎受到机械损伤大而存活率降低;
- 只能敲入外源基因,不能敲除或定点修饰基因;
- 外源基因整合至基因组的效率很低,对经济大动物(猴、牛、羊)的整合效率更低;
- 外源基因随机整合,或影响内源基因本身的表达;
- 对经济大动物,由于需要大量的供体和受体,耗时长、费用昂贵,受到很大的限制。
胚胎干细胞法
胚胎干细胞(embryonic stem cell,ES)是从早期胚胎内细胞团(ICM)分离出来的,能在体外培养的一种高度未分化的多能干细胞。
- 它是一种含正常二倍体染色体的具有发育全能性的细胞。
- 可以在体外进行人工培养,扩增,并能以克隆的形式保存。
ESC 法是将外源 DNA 定向整合到内源基因组中表达。将转染的胚胎干细胞注射入受体囊胚腔,可参与嵌合体的形成,将来出生的动物的生殖系统就有可能整合上外源基因,通过杂交繁育得到纯合目的基因的个体,即为转基因动物。
胚胎干细胞法制备转基因动物的优点:
- 不受细胞代次影响
- 只需将外源基因导入干细胞
- 可以通过筛选细胞提高转基因效率
胚胎干细胞法制备转基因动物的缺点:
- 大动物胚胎干细胞建系困难;
- 第一代动物为嵌合体;
- 转基因周期时间长,效率较低。
反转录病毒感染法
利用反转录病毒的高效率感染和在宿主细胞DNA上的高度整合特性,可以大大提高基因转移的效率。因此可以利用反转录病毒作为目的基因的载体,通过感染实现基因转移,产生嵌合体动物,再经过杂交,筛选即可获得转基因动物。
逆转录病毒感染法优点:在各种转基因方法中,逆转录病毒感染法的转基因效率是最高的,且为单拷贝随机整合。
逆转录病毒感染法缺点:逆转录病毒载体在设计时虽然缺失了病毒的复制功能序列,但是复制大量载体DNA所需的辅助病毒基因组内有可能与目的基因一起整合到同一细胞核中,就可能大量复制病毒,造成严重的污染,故不安全。此外,逆转录病毒载体容量有限,只能转移小片段 DNA(≤ 10 kb),而且逆转录病毒长末端重复的甲基化状态常使转基因的表达缺失。
精子载体法
精子吸附 DNA 的方法
精子吸附 DNA 就使目的基因进入精细胞,再通过受精作用把目的基因传给子代动物,获得转基因动物。外源目的基因进入精细胞可通过与精子共育法、电穿孔导入法和脂质体转染法。
受精技术
精子在吸附外源性 DNA 后,即可以利用其进行受精,受精的方法及途径随着精子的处理方式及卵来源不同而有所不同:
- 人工授精
- 壶腹部手术授精
- 体外授精
精子载体法制备转基因动物的优点:
- 简便易行;
- 耗费低;
- 转染率高;
- 适应性广。
精子载体法制备转基因动物的缺点:
- 不够稳定;
- 随机整合率高。
体细胞核移植法
体细胞核移植法制备转基因动物的优点:
- 适用于大多数物种
- 试验周期短
- 可在细胞水平检测外源基因是否整合
- 细胞核供体容易获得、操作从容
体细胞核移植法制备转基因动物的缺点
- 操作程序相对复杂;
- 可能出现生理缺陷;
- 克隆效率较低。
转基因动物的应用
转基因动物在生命科学基础研究中的应用
- 研究基因的结构与功能
- 研究基因的组织特异性表达
- 研究发育相关基因的表达与调控
- 克隆在发育中起重要作用的基因
- 基因多级调节系统的研究
在农牧业生产上的应用
- 优良动物品种育种
- 快速生长与肉质改良
- 增强抗病性
- 增强抗逆性
转基因动物在医药研究领域中的应用
- 研究病毒性疾病
- 研究建立人类疾病的转基因动物模型
- 转基因动物与基因治疗
- 生产天然活性药物蛋白
在异种器官移植中的应用
建立异种抗原缺失或者表达水平低下的转基因动物(猪)
转基因动物研究及其产品的安全性评价
供体动物和受体动物
- 提供配子或胚胎干细胞的动物(供体)以及受体的动物需要有详细的履历,包括受体、供体的背景资料、生物学特性、生态环境、健康状
况及其他系谱资料等。 - 动物的健康状况应由兽医专家进行评估,包括与物种及繁衍有关疾病问题的特定检查。
- 为控制某些偶发性病原菌的传播,供体和受体动物应满足同样的建立生产群所要求的偶发性病原菌检查标准。而作为异种移植供体使用的转基因动物,对其供体和受体动物来源地区的传染病监测更为严格
- 对工作人员的健康状况也要做系统检查,甚至要终生监测
基因操作的安全性
为确保终产物具有预期性,必须对用来生产转基因动物的重组 DNA 构件加以很好的鉴定,转基因构件的组装、克隆、纯化及最后的鉴定均应有质
量监控。主要包括:
- 转基因及其表达系统(功能的异源获得)
- 通过同源重组实现基因的定位整合(功能的同源丧失)
- 转基因及其表达产物的安全性鉴定
转基因动物释放的安全性评价
(一)遗传稳定性
外源 DNA 插入到宿主动物的基因组中往往牵涉到多拷贝的 DNA 同时整合到染色体的同一位点上,也可能整合位点多于一个,以及整合过程中或整合后,全部或有些基因发生重排或缺失。在传代过程中可能发生染色体交换、易位等引起转基因在基因组中的位置或基因结构的变化,进而引起编码的蛋白发生变化以及新基因的灭活或基因沉默等。因此,转基因动物释放前,其稳定性应该通过诸如 Southern 等方法来检测几代转基因动物的稳定性。
(二)表达稳定性
- 转基因表达的稳定性会发生改变,这种改变取决于宿主动物的遗传背景与转基因之间互作的程度,以及转基因由于父系或母系遗传所表现出的印记作用。有结果表明,随着传递代数增加,转基因表达水平会降低。
- 因此,在一个世代中或经过若于代后, 转基因的稳定性应从表达量确定,应确定一个可接受的表达量的范围作为标准。可能时,正常的或预期的转基因RNA转录水平还应包括转录物的大小、相对丰度、RNA 生成的组织和细胞系等角度进行验证。方法包括 Northern、RT-PCR 等技术。
- 对预期产物的量应从多个转基因动物系谱中检测。
生态环境安全问题
- 以抗病性改良为目标的生产性状改良型转基因动物,其抗病性实际上为病原提供了更强的选择压力,造成病原可能在选择压力下进化的问题,病原的进化不仅对转基因动物带来危害,同时也对非转基因群体带来极大的威胁。
- 转基因动物一旦发生逃逸有可能破获生态平衡。
- 转基因动物应严格隔离,繁育应仅限于特定机构中进行,同时要对转基因动物群体和个体进行监测。
几个转基因动物伦理学方面的问题
- 我们没有权利因为好奇或者其它奇怪的目的干涉生物的基因组
- 在制造转基因动物的过程中有较高的死亡率,为了获得一个成功的转基因动物牺牲那么多的动物是否值得
- 转基因的动物通常是怪物一样的东西,它们要么因为所转的基因寿命缩短,如果它们逃逸出去对于周围环境中的生物会造成威胁。
- 小鼠之类的动物与人类是如此不同,它们怎能成为精准的人类疾病模型
第十章 酵母基因工程
发展历程
- 1974 年,rlarck—walker 和 Miklos 发现在大多数酿酒酵母中存在质粒。
- 1978 年,Hmnen 将来自一株酿酒酵母的 leu 2 基因导入另一株酿酒酵母,弥补了后者的 Leu2 缺陷,标志着酵母重组技术的建立。
- 1981 年,Hinnen 等用酵母基因表达系统表达了人干扰素。
- 1983 年,中国首次用酵母菌表达了乙型肝炎病毒表面抗原基因。
- 1989 年,酵母双杂交系统被用来研究蛋白质间的互作。
- 1996年,在全世界科学家的通力合作下,完成了第一个真核生物——酿酒酵母全基因组的测序。
酵母菌作为表达外源基因受体菌的特征
酵母菌的简介:酵母菌(Yeast)是一群以芽殖或裂殖等无性繁殖或以减数分裂实现有性繁殖。至少由80个属和600多个种组成。
酵母菌是最成熟的真核生物表达系统。
酵母菌表达外源基因的优势:
- 酵母菌是最简单的真核模式生物
- 全基因组测序,基因表达调控机理比较清楚,遗传操作简便
- 具有原核细菌无法比拟的真核蛋白翻译后加工系统
- 大规模发酵历史悠久、技术成熟、工艺简单、成本低廉
- 能将外源基因表达产物分泌至培养基中
- 采用高表达的启动子,如 MOX、AOX、LAC4 等
- 不含有特异性的病毒、不产内毒素,美国 FDA 认定为安全的基因工程受体系统(Generally Recognized As Safe GRAS)
酵母菌表达外源基因的劣势:
- 与原核相比,表达效率相对低,因为表达的外源蛋白会形成聚合体而影响产率
- 酵母常有密码子偏性,真核基因在其中表达时需要人工修正。
酵母菌的宿主系统
广泛用于外源基因表达的酵母宿主菌
目前已广泛用于外源基因表达和研究的酵母菌包括:
- 酿酒酵母,农业、食品,如啤酒、酒精
- 巴斯德毕赤酵母,人血清清蛋白、溶菌酶等
- 粟酒裂殖酵母,凝血因子、细胞周期调节蛋白等
- 多形汉逊酵母,乙型肝炎表面抗原、葡萄糖淀粉酶等
- 乳酸克鲁维酵母,牛凝乳酶、人血清清蛋白、人溶菌酶等
其中酿酒酵母的遗传学和分子生物学研究最详尽,但巴斯德毕赤酵母表达外源基因最理想
提高重组蛋白表达产率的突变宿主菌
- 开发高效的启动子提高和控制外源基因的转录水平
- 提供表达载体在细胞的稳定性
- 提高表达基因在细胞中的拷贝数
抑制超糖基化作用的突变宿主菌
许多真核生物的蛋白质在其天门冬酰胺侧链上接有寡糖基团,它们常常影响蛋白质的生物活性。整个糖单位由糖基核心和外侧糖链两部分组成。
酵母菌普遍拥有蛋白质的糖基化系统,但野生型酿酒酵母对异源蛋白的糖基化反应很难控制,呈超糖基化倾向,因此超糖基化缺陷株非常重要。
减少泛素依赖型蛋白降解作用的突变宿主菌
真核生物如何识别和选择性降解蛋白质是细胞生命过程中的重要环节,对于维持蛋白质在细胞内含量的动态平衡起着关键性作用。需要清除的蛋白质,通过其赖氨酸残基侧链 ε-氨基连接多聚泛素链(降解标签),继而在蛋白酶体中被降解。
泛素-蛋白酶体系统与蛋白质质量控制、细胞周期、DNA 修复、转录及免疫应激等密切相关,也与许多种疾病的发生相关。
泛素降解途径衰减的酿酒酵母 UBI 4 缺陷型:
在酿酒酵母菌中,泛素主要由 UBI 4基因表达,UBI 4- 突变株能正常生长,但细胞内游离泛素分子的浓度比野生株要低得多,因此 UBI 4缺陷突变株是外源基因表达理想的受体。
酵母菌的载体系统
酿酒酵母/穿梭质粒
- 靶基因克隆
- TA 克隆
- 重组表达载体
- 导入酵母
- 诱导表达
- 蛋白纯化
毕赤酵母/整合质粒
- 靶基因克隆
- TA 克隆
- 重组表达载体
- 重组质粒线性化
- 电转化导入酵母
- 诱导表达
- 蛋白纯化
酵母菌的转化系统
酵母菌的转化方法
酵母菌原生质体转化法:早期酵母菌的转化都采用在等渗缓冲液中稳定的原生质体转化,在Ca2+ 和 PEG 的存在下,转化细胞可达原生质体总数的 1 ~ 2%。但该程序操作周期长,而且转化效率受到原生质再生率的严重制约。
碱金属离子介导的酵母菌完整细胞的转化:酿酒酵母的完整细胞经碱金属离子(如 Li+ 等)、PEG、热休克处理后,也可高效吸收质粒 DNA,而且具有下列特性:
- 吸收线型 DNA 的能力明显大于环状 DNA,两者相差 80 倍
- 共转化现象极为罕见
酵母菌电击转化法:酵母菌原生质体和完整细胞均可在电击条件下吸收质粒 DNA,但在此过程中应避免使用 PEG,它对受电击的细胞具有较很大的负作用。电击转化的优点是不依赖于受体细胞的遗传特征及培养条件适用范围广,而且转化率可高达 105 /μg DNA。
用于转化子筛选的标记基因
用于酵母菌转化子筛选的标记基因主要有营养缺陷型互补基因和显性基因两大类
营养缺陷型的互补基因主要有氨基酸和核苷酸生物合成基因,如:LEU、TRP、HIS、LYS、URA、ADE
显性标记基因编码产物只要是毒性物质的抗性蛋白:
| 标记基因 | 编码产物 | 遗传表型 |
|---|---|---|
| aph | 氨基糖苷转移酶 | 抗 G418 |
| cat | 氯霉素乙酰转移酶 | 抗氯霉素 |
| dhfr | 二氢叶酸还原酶 | 抗氨甲喋呤和磺胺 |
| cup1 | 铜离子螯合物 | 耐受铜离子 |
| suc2 | 蔗糖转化酶 | 耐受高浓度蔗糖 |
| ilv2 | 乙酰乳糖合成酶 | 抗硫酰脲除草剂 |
酵母菌的表达系统
酵母菌启动子的可控性
超诱导型启动子
酿酒酵母的半乳糖利用酶系由 GAL1、GAL7和 GAL10 基因编码
半乳糖诱导时,GAL4 高效表达,GAL1、GAL1、GAL10 超高效表达
外源基因在酵母菌中表达的限制因素
- 外源基因稳态 mRNA 的浓度
- 外源基因 mRNA 的翻译活性
- 酵母菌对密码子的偏爱性
在酿酒酵母中,高丰度的蛋白质(如甘油醛-3-磷酸脱氢酶 GAPDH、磷酸甘油激酶 PKG、乙醇脱氢酶 ADH)中 96% 以上的氨基酸是由 25 个密码子编码的
酵母菌表达系统的选择
(一)酿酒酵母表达系统
酿酒酵母的基因表达系统最为成熟,包括转录活性较高的甘油醛-3-磷酸脱氢酶基因 GAPDH、磷酸甘油激酶基因 PKG、乙醇脱氢酶基因 ADH 所属的启动子,多种重组外源蛋白获得成功表达。
酿酒酵母表达系统的最大问题在于其超糖基化能力,往往使得有些重组蛋白(如人血清白蛋白等)与受体细胞紧密结合,而不能大量分泌。这一缺陷可用非酿酒酵母型的表达系统来弥补。
(二)乳酸克鲁维酵母表达系统
乳酸克鲁维酵母的双链环状质粒 pKD1 已被广泛用作重组异源蛋白生产的高效表达稳定性载体,即便在无选择压力的条件下,也能稳定遗传 40 代以上。
乳酸克鲁维酵母表达分泌型和非分泌型的重组蛋白,性能均优于酿酒酵母表达系统。
(三)巴斯德毕赤酵母表达系统
巴斯德毕赤酵母是一种甲基营养菌,能在低廉的甲醇培养基中生长,甲醇可高效诱导甲醇代谢途径中各酶编码基因的表达,因此生长迅速、乙醇氧化酶基因 AOX1 所属强启动子、表达的可诱导性是巴斯德毕赤酵母表达系统的三大优势。
由于巴斯德毕赤酵母没有合适的自主复制型载体,所以外源基因的表达序列一般整合入受体的染色体DNA上。在此情况下,外源基因的高效表达在很大程度上取决于整合拷贝数的多寡。目前已有百余种具有经济价值的重组蛋白在巴斯德毕赤酵母系统中获得成功表达。
(四)多形汉逊酵母表达系统
多形汉逊酵母也是一种甲基营养菌。其自主复制序列 HARS 已被克隆,并用于构建克隆表达载体,但与巴斯德毕赤酵母相似,这种载体在受体细胞有丝分裂时显示出不稳定性。所不同的是,HARS 质粒能高频自发地整合在受体的染色体 DNA 上,甚至可以连续整合 100 多个拷贝,因此重组多形汉逊酵母的构建也是采取整合的策略。
目前,包括乙型肝炎表面抗原在内的数种外源蛋白在该系统中获得成功表达。
酵母基因工程的应用
利用重组酵母生产乙肝疫苗
由乙型肝炎病毒(HBV)感染引起的急慢性乙型肝炎是一种严重的传染病,全球有 3 亿人成为 HBV 携带者,中国就有 1.2 亿,占 1/9;相当一部分人可能转化为肝硬化或肝癌患者。目前对乙型肝炎病毒还没有一种有效的治疗药物,因此高纯度乙型疫苗的生产对预防病毒感染具有重大的社会效益,而利用重组酵母大规模生产乙型疫苗为其广泛应用提供了可靠的保证。
(一)乙型肝炎病毒的结构与性质
乙型肝炎病毒的结构:乙肝病毒是一种蛋白包裹型的双链 DNA 病毒,具有感染力的病毒颗粒呈球面状,直径为 42 nm,基因组仅为 3.2 kb。病毒颗粒的主要结构蛋白是病毒的表面抗原多肽(HBsAg)或 S 多肽,它具有糖基化和非糖基化两种形式。颗粒内的蛋白成份包括核心抗原( HBcAg )、病毒 DNA 聚合酶、微量病毒蛋白。除此之外,被乙肝病毒感染的人的肝脏还能合成并释放大量的 22 nm 的空壳亚病毒颗粒,其免疫原性是未装配的各种包装蛋白组份的 1000 倍。包装蛋白共有三种转膜糖蛋白:S、M、L 多肽。
传统乙肝疫苗的制备
乙肝病毒在体外细胞培养基中并不能繁殖,因此第一代的乙肝疫苗是从病毒携带者的肝细胞质膜上提取出来的。虽然这种质膜来源的疫苗具有较高的免疫原性,但由于原材料的限制难以大规模产业化。
(二)产乙肝表面抗原的重组酿酒酵母
20 世纪 80 年代开始选择酿酒酵母表达重组 HBsAg,主要工作包括将 S 多肽的编码置于 ADH1 启动子控制下,转化子能表达出具有免疫活性的重组蛋白,它在细胞提取物中以球形脂蛋白颗粒的形式存在,平均颗粒直径为 22 nm,其结构和形态均与慢性乙肝病毒携带者血清中的病毒颗粒相同。
目前,由酿酒酵母生产的重组 HBsAg 颗粒已作为乙肝疫苗商品化重组产物的最终产量可达细胞总蛋白量的 1% ~ 2%。
进一步的研究表明,M 多肽和 L 多肽对 S 型疫苗具有显著的增效作用,由三者(或两者)构成的复合型乙肝疫苗还可以诱导那些对 S 抗原缺乏响应的人群的免疫反应。
(三)产乙肝表面抗原的重组巴斯德毕赤酵母
重组巴斯德毕赤酵母的性能:由于巴斯德毕赤酵母染色体 DNA 上还拥有第二个乙醇氧化酶基因 AOX2,所以整合型重组菌仍能在含有甲醇的培养基上生长。重组菌首先在含有甘油的培养基中培养,待甘油耗尽后,加入甲醇诱导 HBsAg 表达,最终 S 蛋白的产量可达细胞可溶性蛋白总量的 3%,在大规模的生产过程中,巴斯德毕赤酵母工程菌在一个 240 L 的发酵罐中培养,最终可获得 90 克 22 nm 的 HBsAg 颗粒,足够制成 900 万份乙肝疫苗。
利用重组酿酒酵母提高酿酒工艺
- 将葡萄糖淀粉酶基因导入酿酒酵母;
- 将外源的蛋白水解酶基因导入酿酒酵母;
- 将 β-葡聚糖酶基因导入酵母;
- 将 ATP 硫酸化酶和腺苷酰硫酸激酶基因在酿酒酵母体内表达,促进 SO2 的生成,维持啤酒、葡萄酒等风味。
利用重组酿酒酵母环境能源危机
通过转基因技术使酿酒酵母能利用纤维素和半纤维素等可再生物质来生产廉价的酒精,缓解能源紧张。
酵母基因工程在生物医药中的应用
- 生物反应器。人血清白蛋白、溶菌酶、乙肝疫苗等。
- 人类蛋白质间的互作。如利用酵母表达系统开展分泌蛋白和受体基因的快速筛选。
- 筛选新药。利用酵母展示技术筛选细胞凋亡的抑制剂、HIV-1 整合抑制新要等。
- 利用酵母表达膜蛋白。
第十一章 基因治疗
基因药物
基因工程药物包括蛋白质类药物和核酸类药物两类:
- 蛋白质类药物主要有重组细胞因子、重组多肽类激素、重组溶栓药物、基因工程抗体和基因工程疫苗等。
- 核酸类药物主要有 DNA 药物,反义核酸药和 RNA 干扰药物等。主要针对的疾病有肿瘤、感染性疾病、人免疫缺陷病毒相关疾病、自身免疫性疾病、器官移植、心血管疾病、神经障碍性疾病等。
基因工程药物的作用机理分为以下三类:
- 蛋白或多肽药物:通过蛋白自身的生理生化特性而抵抗疾病,胰岛素、生长素等激素,各类生长因子。
- 基因工程疫苗、基因工程抗体:基于抗原抗体反应的原理来抵抗疾病;乙肝疫苗,新冠肺炎疫苗,霍乱菌疫苗等。
- 反义核酸、核酶和 RNAi,基于中断基因表达来抵抗疾病。福米韦生,第一代反义核酸药物,治疗艾滋病(AIDS)病人并发的巨细胞病毒(CMV)性视网膜炎。通过对人类巨细胞病毒(CMV)mRNA 的反义抑制发挥特异而强大的抗病毒作用。
基因诊断
基因诊断常用技术方法如下:
- 核酸分子杂交技术
- 聚合酶链反应 (PCR)
- 基因测序
- 生物芯片
临床治疗
传统临床疾病治疗有以下四种主要方式:
- 药物治疗
- 手术治疗
- 放射治疗
- 理疗
新的生物治疗:基因治疗 (gene therapy)
基因治疗的概念
- 经典概念(狭义概念):针对患者的基因缺陷,导入特定基因补偿其缺陷或赋予机体新的功能以抗衡缺陷。
- 广义概念:将目的基因放进特定载体中导入靶细胞或组织,通过替换或补偿引起疾病的基因,调节异常表达的基因来治疗或缓解疾病,均称之为基因治疗。
基因治疗的总体策略
- 基因矫正
- 基因置换
- 基因增补
- 基因失活
- 自杀基因的应用
- 免疫基因治疗
- 耐药基因治疗
基因矫正 (gene correction) 是指将致病基因的突变碱基加以纠正,而保留正常部分。
基因置换 (gene replacement)指用正常基因通过同源重组技术,原位替换致病基因,使细胞内的 DNA 完全恢复正常状态。
基因增补 (gene augmentation)是指不去除异常基因,将有功能的正常基因导入病变细胞或其它细胞后发生非定点整合,表达正常产物以补偿缺陷基因的功能,或使原有的功能得以加强。
基因失活 (gene inactivation) 是将特定的反义核酸(反义 RNA、反义 DNA)和核酶导入细胞,在转录和翻译水平阻断某些基因的表达,而实现治疗的目的。多用于抗肿瘤和抗病毒感染的治疗。
“自杀基因”的应用:HSV-TK
自杀基因又称前体药物酶转化基因,这种基因导入受体细胞后可产生一种酶,它可将原来无细胞毒性或低毒药物前体转化为细胞毒性物质,将细胞本身杀死,这种基因被称为“自杀基因”。
自杀基因导入肿瘤细胞后,可将肿瘤细胞杀死。但对正常细胞则无伤害作用。
免疫基因治疗:是把产生抗病毒或者肿瘤免疫力对应的抗原决定族基因导入机体细胞,以达到治疗目的。如细胞因子基因治疗 、免疫增强基因疗法、肿瘤DNA疫苗疗法等。
耐药基因治疗:在肿瘤治疗中,为提高机体耐受肿瘤化疗药物的能力,把产生抗药物毒性的基因导入人体细胞。如将多药耐药基因 MDR-1 导入骨髓造血干细胞,减少骨髓受抑制的程度,以加大化疗剂量,提高化疗效果。
基因治疗的方式
根据基因导入的方式分为两种:
- 直接体内疗法(in vivo)是指将目的基因直接导入体内有关的组织器官,使其进入相应的细胞并进行表达。
- 间接体内疗法(ex vivo)是指在体外将目的基因导入靶细胞,经过筛选和增殖后将细胞回输给患者,使该基因在体内有效地表达相应产物,以达到治疗的目的。
基因治疗的基本程序
- 治疗性基因的获得
- 基因载体的选择,病毒载体、非病毒载体
- 靶细胞的选择,体细胞、生殖细胞
- 基因转移方法,化学法、物理法、病毒载体系统、膜融合法等
- 转导细胞的选择鉴定,利用载体中的标记基因
- 回输体内
(一)治疗性基因的获得
外源基因
目的基因:
- 与致病基因相对应的有功能的正常基因
- 与致病基因无关、有治疗作用的基因
标记基因:新霉素磷酸转移酶(Neo)基因
外源基因的获得:人工合成、PCR 扩增
外源基因的选择原则:
- 体内少量表达就可显著改善症状;
- 过高表达不会对机体造成危害;
- 在抗病毒和病原体的基因治疗中,所选择的靶基因应在病毒和病原体的生活史中起重要的作用,并且该序列是特异的;肿瘤病人多有免疫缺陷,可选用免疫因子基因转入人体;可采用反义技术封闭细胞内活化的癌基因或向细胞内转入野生型抑癌基因,抑制肿瘤生长,所针对的癌基因或抑癌基因应与该肿瘤的发生和发展有明确的相关性。
(二)载体的选择与构建
基因治疗关键步骤之一,是将治疗基因高效转移入患者体内、并能调控其适度表达。
病毒载体:
- 逆转录病毒(RV)
- 腺病毒(AV)
- 腺相关病毒(AAV)
- 单纯疱疹病毒(HSV)
- 痘苗病毒(VV)
问题:
- 靶细胞 DNA 合成必须活跃,因为病毒感染和整合作用均依赖靶细胞 DNA 的复制;
- 安全性问题。随机整合:诱发插入突变,激活癌基因;缺陷型逆转录病毒通过重组获得复制能力
(三)靶细胞的选择
间接体内疗法中使用,分为生殖细胞和体细胞两大类,现阶段只限于应用体细胞
靶细胞选择的原则:
- 易操作;
- 易培养;
- 易接受外源基因;
- 在体内能持久高效表达
目前常用的靶细胞包括:成纤维细胞、淋巴细胞、上皮细胞、造血细胞、肝细胞、肌细胞等
生殖细胞基因治疗:
- 生殖生物学问题复杂
- 伦理学问题
- 禁止应用人体
体细胞基因治疗:研究重点
(四)基因转移方法
- 化学方法——磷酸钙沉淀法、DEAE-葡聚糖法
- 物理方法——电穿孔法、粒子轰击法
- 病毒法——主要通过携带有外源基因的病毒载体感染靶细胞来实现基因的转移。
- 膜融合法——细胞融合法、脂质体介导法、原生质体融合法、微细胞核介导法等
(五) 基因转染细胞的筛选与鉴定
抗性筛选方法:重组载体上插入有标记基因 Neo,导入受体细胞后可使其产生对 G418(氨基糖甙抗生素)药物的抗性。在加入 G418 的培养基中进行选择性培养时,转染 NeoR 基因的细胞能够存活,而未转染的细胞则死亡;
应用标记基因作为探针进行分子杂交法筛选
外源基因表达的鉴定:采用 Northern 印迹杂交;检测 mRNA 的表达;测定其表达产物即蛋白质的含量等方法。
(六)回输体内
方式:静脉注射、肌注、皮下注射、滴鼻等
- 基因修饰的淋巴细胞以静脉注射的方式回输到血液中;
- 将皮肤成纤维细胞以细胞胶原悬液注射至患者皮下组织;
- 采用自体骨髓移植的方法输入造血细胞;
- 或以导管技术将血管内皮细胞定位输入血管等。
基因治疗的现状与展望
基因治疗应满足的条件:
- 优先单基因缺陷疾病
- 仅限体细胞的基因治疗
- 靶细胞易获取、培养、及回输体内。
- 治疗效果应胜过对病人的危害。
- 表达水平无需调控且无副作用。
- 需经动物实验验证安全可行。
基因治疗的合理性:
- 基因治疗的疾病一般是目前无法得到根治的、严重的遗传性疾病,而且这些疾病很容易造成患者死亡,因此,基因治疗方案比较容易得到患者或监护人的同意,同时也比较容易得到政府有关部门的批准。
- 尽管基因治疗困难重重,但是科学家认为,基因治疗的思路是正确的,因此一直在坚持不懈地研究着、实践着。
- 基因治疗蕴藏着丰厚的商业利润。因此在巨大商业利润的驱动下,国家或私人都投入了大量研究资金,这样就进一步刺激了基因治疗的发展。
基因治疗的困难:
- 难以获得真正有治疗作用的基因。
- 外源基因的表达难以在体内精确调控。
- 体细胞经体外培养后,其生物学特性会有改变。
- 过多的外源蛋白对机体带来可能的影响。
- 随机整合潜在的威胁。
第十二章 转基因生物安全性及安全监管
转基因安全评价原则
实质等同性原则:即生物技术产生的食品及食品成分,如果与一种现有的食物成分、毒理性、过敏性、抗营养因子等在实质上是相当的,则可以认为是安全的。强调了转基因食品安全性的目的,不是要了解该食品的绝对安全性,而是评价它与非转基因食品的同类食品比较的相对安全性。
预先防范原则:采取对公众透明的方式,结合其他的评价原则,对转基因生物及其产品研究和试验进行风险性和危害性评价后采取先预防,防患于未然。
个案原则:针对不同目的基因、不同载体、不同受体、不同释放环境进行个案评价
逐步评估原则:转基因生物及其产品的研究开发包括实验室研究,中间试验,环境释放,生产性试验和商业化生产等环节,该原则就是要求在每个环节上对转基因生物及其产品进行风险评估。
风险效益平衡原则:对转基因生物及其产品的经济效益和它可能给环境和人类健康带来风险进行权衡,从而确定是否继续开发相关产品。
熟悉性原则:转基因生物及其产品的风险评价工作既可以在短期内完成,也可能需要长期监测。这主要取决于人们对转基因生物及其产品的有关性状,同其他生物或环境的相互作用,预定用途等背景知识的熟悉程度等。
转基因安全评价内容
分子水平评价:基因水平评价、转录水平评价、翻译水平评价
食用或饲用安全性评价:营养学评价、毒理性评价、致敏性评价、非预期效应评价
环境安全性评价:基因漂移评价、遗传稳定性评价、生存竞争力评价、生物多样性评价